Pre-trained ODAC models are versatile across various MOF-related tasks. To begin, we’ll start with a fundamental application: calculating the adsorption energy for a single CO2 molecule. This serves as an excellent and simple demonstration of what you can achieve with these datasets and models.
For predicting the adsorption energy of a single CO2 molecule within a MOF structure, the adsorption energy () is defined as:
Each term on the right-hand side represents the energy of the relaxed state of the indicated chemical system. For a comprehensive understanding of our methodology for computing these adsorption energies, please refer to our paper.
Loading Pre-trained Models¶
Need to install fairchem-core or get UMA access or getting permissions/401 errors?
Install the necessary packages using pip, uv etc
! pip install fairchem-core fairchem-data-oc fairchem-applications-cattsunamiGet access to any necessary huggingface gated models
Get and login to your Huggingface account
Request access to https://
huggingface .co /facebook /UMA Create a Huggingface token at https://
huggingface .co /settings /tokens/ with the permission “Permissions: Read access to contents of all public gated repos you can access” Add the token as an environment variable using
huggingface-cli loginor by setting the HF_TOKEN environment variable.
# Login using the huggingface-cli utility
! huggingface-cli login
# alternatively,
import os
os.environ['HF_TOKEN'] = 'MY_TOKEN'A pre-trained model can be loaded using FAIRChemCalculator. In this example, we’ll employ UMA to determine the CO2 adsorption energies.
from fairchem.core import FAIRChemCalculator, pretrained_mlip
predictor = pretrained_mlip.get_predict_unit("uma-s-1p2")
calc = FAIRChemCalculator(predictor, task_name="odac")WARNING:root:device was not explicitly set, using device='cuda'.
Adsorption in rigid MOFs: CO2 Adsorption Energy in Mg-MOF-74¶
Let’s apply our knowledge to Mg-MOF-74, a widely studied MOF known for its excellent CO2 adsorption properties. Its structure comprises magnesium atomic complexes connected by a carboxylated and oxidized benzene ring, serving as an organic linker. Previous studies consistently report the CO2 adsorption energy for Mg-MOF-74 to be around -0.40 eV [1] [2] [3].
Our goal is to verify if we can achieve a similar value by performing a simple single-point calculation using UMA. In the ODAC23 dataset, all MOF structures are identified by their CSD (Cambridge Structural Database) code. For Mg-MOF-74, this code is OPAGIX. We’ve extracted a specific OPAGIX+CO2 configuration from the dataset, which exhibits the lowest adsorption energy among its counterparts.
import matplotlib.pyplot as plt
from ase.io import read
from ase.visualize.plot import plot_atoms
mof_co2 = read("structures/OPAGIX_w_CO2.cif")
mof = read("structures/OPAGIX.cif")
co2 = read("structures/co2.xyz")
fig, ax = plt.subplots(figsize=(5, 4.5), dpi=250)
plot_atoms(mof_co2, ax)
ax.set_axis_off()
The final step in calculating the adsorption energy involves connecting the FAIRChemCalculator to each relaxed structure: OPAGIX+CO2, OPAGIX, and CO2. The structures used here are already relaxed from ODAC23. For simplicity, we assume here that further relaxations can be neglected. We will show how to go beyond this assumption in the next section.
mof_co2.calc = calc
mof.calc = calc
co2.calc = calc
E_ads = (
mof_co2.get_potential_energy()
- mof.get_potential_energy()
- co2.get_potential_energy()
)
print(f"Adsorption energy of CO2 in Mg-MOF-74: {E_ads:.3f} eV")Adsorption energy of CO2 in Mg-MOF-74: -0.473 eV
Adsorption in flexible MOFs¶
The adsorption energy calculation method outlined above is typically performed with rigid MOFs for simplicity. Both experimental and modeling literature have shown, however, that MOF flexibility can be important in accurately capturing the underlying chemistry of adsorption [1] [2] [3]. In particular, uptake can be improved by treating MOFs as flexible. Two types of MOF flexibility can be considered: intrinsic flexibility and deformation induced by guest molecules. In the Open DAC Project, we consider the latter MOF deformation by allowing the atomic positions of the MOF to relax during geometry optimization [4]. The addition of additional degrees of freedoms can complicate the computation of the adsorption energy and necessitates an extra step in the calculation procedure.
The figure below shows water adsorption in the MOF with CSD code WOBHEB with added defects (WOBHEB_0.11_0) from a DFT simulation. A typical adsorption energy calculation would only seek to capture the effects shaded in purple, which include both chemisorption and non-bonded interactions between the host and guest molecule. When allowing the MOF to relax, however, the adsorption energy also includes the energetic effect of the MOF deformation highlighted in green.
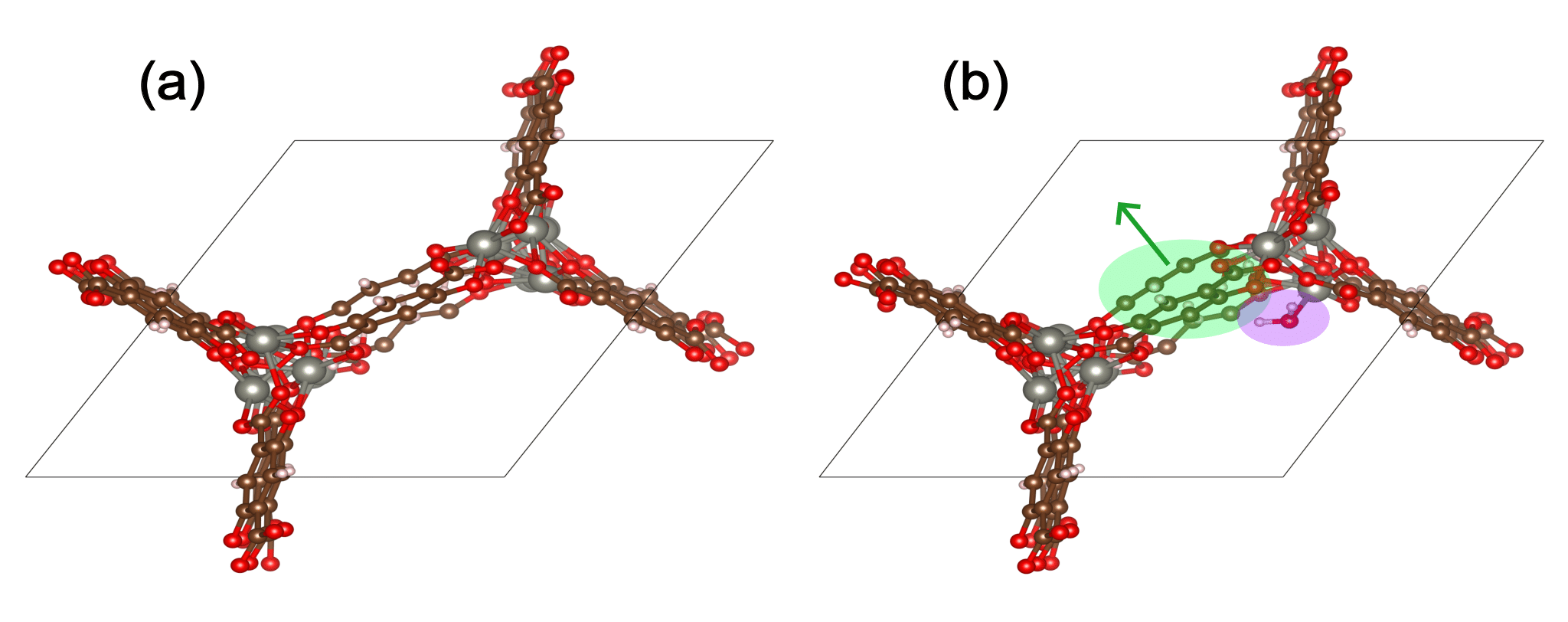
To account for this deformation, it is vital to use the most energetically favorable MOF geometry for the empty MOF term in Eqn. 1. Including MOF atomic coordinates as degrees of freedom can result in three possible outcomes:
The MOF does not deform, so the energies of the relaxed empty MOF and the MOF in the adsorbed state are the same
The MOF deforms to a less energetically favorable geometry than its ground state
The MOF locates a new energetically favorable geoemtry relative to the empty MOF relaxation
The first outcome requires no additional computation because the MOF rigidity assumption is valid. The second outcome represents physical and reversible deformation where the MOF returns to its empty ground state upon removal of the guest molecule. The third outcome is often the result of the guest molecule breaking local symmetry. We also found cases in ODAC in which both outcomes 2 and 3 occur within the same MOF.
To ensure the most energetically favorable empty MOF geometry is found, an addition empty MOF relaxation should be performed after MOF + adsorbate relaxation. The guest molecule should be removed, and the MOF should be relaxed starting from its geometry in the adsorbed state. If all deformation is reversible, the MOF will return to its original empty geometry. Otherwise, the lowest energy (most favorable) MOF geometry should be taken as the reference energy, , in Eqn. 1.
H2O Adsorption Energy in Flexible WOBHEB with UMA¶
The first part of this tutorial demonstrates how to perform a single point adsorption energy calculation using UMA. To treat MOFs as flexible, we perform all calculations on geometries determined by geometry optimization. The following example corresponds to the figure shown above (H2O adsorption in WOBHEB_0.11_0).
In this tutorial, corresponds to the energy of determined from geometry optimization of .
First, we obtain the energy of the empty MOF from relaxation of only the MOF:
import ase.io
from ase.optimize import BFGS
mof = ase.io.read("structures/WOBHEB_0.11.cif")
mof.calc = calc
relax = BFGS(mof)
relax.run(fmax=0.05)
E_mof_empty = mof.get_potential_energy()
print(f"Energy of empty MOF: {E_mof_empty:.3f} eV") Step Time Energy fmax
BFGS: 0 14:48:56 -1077.368915 0.129114
BFGS: 1 14:48:57 -1077.370392 0.075187
BFGS: 2 14:48:57 -1077.372340 0.145326
BFGS: 3 14:48:58 -1077.374554 0.111789
BFGS: 4 14:48:58 -1077.376096 0.074286
BFGS: 5 14:48:58 -1077.377455 0.063780
BFGS: 6 14:49:00 -1077.378941 0.080821
BFGS: 7 14:49:00 -1077.380760 0.096851
BFGS: 8 14:49:01 -1077.382639 0.078402
BFGS: 9 14:49:04 -1077.384446 0.086890
BFGS: 10 14:49:04 -1077.386281 0.083295
BFGS: 11 14:49:05 -1077.388392 0.084063
BFGS: 12 14:49:05 -1077.390737 0.069052
BFGS: 13 14:49:05 -1077.393126 0.076013
BFGS: 14 14:49:06 -1077.395558 0.084328
BFGS: 15 14:49:06 -1077.398145 0.079955
BFGS: 16 14:49:06 -1077.400825 0.079975
BFGS: 17 14:49:07 -1077.403366 0.067396
BFGS: 18 14:49:07 -1077.405675 0.070441
BFGS: 19 14:49:08 -1077.407934 0.087883
BFGS: 20 14:49:08 -1077.410401 0.084001
BFGS: 21 14:49:09 -1077.413125 0.059825
BFGS: 22 14:49:09 -1077.415973 0.071945
BFGS: 23 14:49:09 -1077.418820 0.067802
BFGS: 24 14:49:10 -1077.421568 0.069925
BFGS: 25 14:49:10 -1077.424156 0.067339
BFGS: 26 14:49:11 -1077.426522 0.060834
BFGS: 27 14:49:11 -1077.428607 0.069320
BFGS: 28 14:49:11 -1077.430416 0.060293
BFGS: 29 14:49:12 -1077.431998 0.051494
BFGS: 30 14:49:13 -1077.433389 0.056302
BFGS: 31 14:49:13 -1077.434620 0.057619
BFGS: 32 14:49:14 -1077.435743 0.046081
Energy of empty MOF: -1077.436 eV
Next, we add the H2O guest molecule and relax the MOF + adsorbate to obtain .
mof_h2o = ase.io.read("structures/WOBHEB_H2O.cif")
mof_h2o.calc = calc
relax = BFGS(mof_h2o)
relax.run(fmax=0.05)
E_combo = mof_h2o.get_potential_energy()
print(f"Energy of MOF + H2O: {E_combo:.3f} eV") Step Time Energy fmax
BFGS: 0 14:49:14 -1091.661287 1.120236
BFGS: 1 14:49:15 -1091.679631 0.313939
BFGS: 2 14:49:16 -1091.683945 0.232091
BFGS: 3 14:49:16 -1091.695507 0.302372
BFGS: 4 14:49:16 -1091.701044 0.210321
BFGS: 5 14:49:18 -1091.707226 0.171311
BFGS: 6 14:49:18 -1091.712983 0.183136
BFGS: 7 14:49:20 -1091.720515 0.262570
BFGS: 8 14:49:20 -1091.727865 0.202838
BFGS: 9 14:49:21 -1091.735396 0.175191
BFGS: 10 14:49:22 -1091.743447 0.214466
BFGS: 11 14:49:23 -1091.752655 0.253300
BFGS: 12 14:49:23 -1091.762637 0.232873
BFGS: 13 14:49:24 -1091.773118 0.197326
BFGS: 14 14:49:24 -1091.784458 0.164083
BFGS: 15 14:49:25 -1091.796073 0.252840
BFGS: 16 14:49:26 -1091.806471 0.270276
BFGS: 17 14:49:26 -1091.815238 0.186126
BFGS: 18 14:49:27 -1091.822965 0.130935
BFGS: 19 14:49:27 -1091.830272 0.120407
BFGS: 20 14:49:27 -1091.837491 0.140917
BFGS: 21 14:49:28 -1091.844729 0.154729
BFGS: 22 14:49:28 -1091.851965 0.162441
BFGS: 23 14:49:29 -1091.858807 0.165891
BFGS: 24 14:49:30 -1091.864202 0.170328
BFGS: 25 14:49:31 -1091.868721 0.411585
BFGS: 26 14:49:31 -1091.873930 0.221378
BFGS: 27 14:49:32 -1091.880144 0.092263
BFGS: 28 14:49:32 -1091.884364 0.091139
BFGS: 29 14:49:32 -1091.889064 0.135339
BFGS: 30 14:49:33 -1091.893586 0.143430
BFGS: 31 14:49:33 -1091.899476 0.231294
BFGS: 32 14:49:33 -1091.904615 0.312374
BFGS: 33 14:49:34 -1091.908880 0.329443
BFGS: 34 14:49:35 -1091.914106 0.200617
BFGS: 35 14:49:36 -1091.920894 0.173357
BFGS: 36 14:49:36 -1091.927119 0.178003
BFGS: 37 14:49:37 -1091.934498 0.313601
BFGS: 38 14:49:37 -1091.939736 0.149114
BFGS: 39 14:49:40 -1091.943184 0.666278
BFGS: 40 14:49:40 -1091.951219 0.202156
BFGS: 41 14:49:41 -1091.958015 0.134687
BFGS: 42 14:49:41 -1091.968704 0.231915
BFGS: 43 14:49:42 -1091.977391 0.308383
BFGS: 44 14:49:42 -1091.989159 0.196087
BFGS: 45 14:49:43 -1091.995971 0.884631
BFGS: 46 14:49:44 -1092.009727 0.527852
BFGS: 47 14:49:45 -1092.025016 0.196672
BFGS: 48 14:49:45 -1092.048945 0.583762
BFGS: 49 14:49:46 -1092.066870 0.473106
BFGS: 50 14:49:46 -1092.082180 1.045588
BFGS: 51 14:49:46 -1092.106965 0.400884
BFGS: 52 14:49:47 -1092.125492 0.345249
BFGS: 53 14:49:47 -1092.147880 0.349071
BFGS: 54 14:49:48 -1092.159613 0.423525
BFGS: 55 14:49:51 -1092.173599 0.387529
BFGS: 56 14:49:51 -1092.197354 0.316005
BFGS: 57 14:49:52 -1092.207718 0.284365
BFGS: 58 14:49:52 -1092.226776 0.288696
BFGS: 59 14:49:53 -1092.236144 0.357350
BFGS: 60 14:49:53 -1092.248636 0.306531
BFGS: 61 14:49:54 -1092.258910 0.274180
BFGS: 62 14:49:54 -1092.267432 0.161252
BFGS: 63 14:49:55 -1092.273004 0.132088
BFGS: 64 14:49:55 -1092.278970 0.121835
BFGS: 65 14:49:55 -1092.284636 0.124332
BFGS: 66 14:49:56 -1092.289819 0.139735
BFGS: 67 14:49:56 -1092.294424 0.169707
BFGS: 68 14:49:56 -1092.298884 0.130593
BFGS: 69 14:49:57 -1092.303164 0.134850
BFGS: 70 14:49:57 -1092.307246 0.133646
BFGS: 71 14:49:57 -1092.311313 0.144179
BFGS: 72 14:49:58 -1092.315626 0.170086
BFGS: 73 14:49:58 -1092.319998 0.162307
BFGS: 74 14:49:58 -1092.323908 0.133380
BFGS: 75 14:49:59 -1092.327224 0.094680
BFGS: 76 14:50:00 -1092.330245 0.129299
BFGS: 77 14:50:00 -1092.332990 0.108176
BFGS: 78 14:50:00 -1092.335153 0.074998
BFGS: 79 14:50:02 -1092.336739 0.067135
BFGS: 80 14:50:03 -1092.338065 0.059953
BFGS: 81 14:50:04 -1092.339383 0.066429
BFGS: 82 14:50:04 -1092.340754 0.074828
BFGS: 83 14:50:04 -1092.342141 0.081612
BFGS: 84 14:50:05 -1092.343542 0.086574
BFGS: 85 14:50:06 -1092.344934 0.092773
BFGS: 86 14:50:06 -1092.346289 0.085472
BFGS: 87 14:50:07 -1092.347557 0.055385
BFGS: 88 14:50:07 -1092.348698 0.043692
Energy of MOF + H2O: -1092.349 eV
We can now isolate the MOF atoms from the relaxed MOF + H2O geometry and see that the MOF has adopted a geometry that is less energetically favorable than the empty MOF by ~0.2 eV. The energy of the MOF in the adsorbed state corresponds to .
mof_adsorbed_state = mof_h2o[:-3]
mof_adsorbed_state.calc = calc
E_mof_adsorbed_state = mof_adsorbed_state.get_potential_energy()
print(f"Energy of MOF in the adsorbed state: {E_mof_adsorbed_state:.3f} eV")Energy of MOF in the adsorbed state: -1077.150 eV
H2O adsorption in this MOF appears to correspond to Case #2 as outlined above. We can now perform re-relaxation of the empty MOF starting from the geometry.
relax = BFGS(mof_adsorbed_state)
relax.run(fmax=0.05)
E_mof_rerelax = mof_adsorbed_state.get_potential_energy()
print(f"Energy of re-relaxed empty MOF: {E_mof_rerelax:.3f} eV") Step Time Energy fmax
BFGS: 0 14:50:08 -1077.149992 1.015996
BFGS: 1 14:50:08 -1077.191180 0.891448
BFGS: 2 14:50:08 -1077.242261 0.659166
BFGS: 3 14:50:09 -1077.289501 0.488322
BFGS: 4 14:50:09 -1077.307292 0.357585
BFGS: 5 14:50:11 -1077.323951 0.300661
BFGS: 6 14:50:11 -1077.337914 0.323515
BFGS: 7 14:50:12 -1077.350368 0.260238
BFGS: 8 14:50:13 -1077.357629 0.135732
BFGS: 9 14:50:14 -1077.362431 0.127846
BFGS: 10 14:50:14 -1077.366942 0.177610
BFGS: 11 14:50:15 -1077.371687 0.171239
BFGS: 12 14:50:16 -1077.376450 0.137789
BFGS: 13 14:50:16 -1077.381090 0.125114
BFGS: 14 14:50:17 -1077.385634 0.149879
BFGS: 15 14:50:17 -1077.389560 0.127104
BFGS: 16 14:50:18 -1077.392628 0.091486
BFGS: 17 14:50:20 -1077.395290 0.089489
BFGS: 18 14:50:21 -1077.397968 0.113624
BFGS: 19 14:50:23 -1077.400667 0.114600
BFGS: 20 14:50:23 -1077.403348 0.105309
BFGS: 21 14:50:24 -1077.405987 0.100961
BFGS: 22 14:50:24 -1077.408531 0.085584
BFGS: 23 14:50:24 -1077.410879 0.095105
BFGS: 24 14:50:25 -1077.412990 0.075064
BFGS: 25 14:50:25 -1077.414890 0.078715
BFGS: 26 14:50:26 -1077.416601 0.078687
BFGS: 27 14:50:26 -1077.418058 0.079494
BFGS: 28 14:50:27 -1077.419281 0.049146
Energy of re-relaxed empty MOF: -1077.419 eV
The MOF returns to its original empty reference energy upon re-relaxation, confirming that this deformation is physically relevant and is induced by the adsorbate molecule. In Case #3, this re-relaxed energy will be more negative (more favorable) than the original empty MOF relaxation. Thus, we take the reference empty MOF energy ( in Eqn. 1) to be the minimum of the original empty MOF energy and the re-relaxed MOf energy:
E_mof = min(E_mof_empty, E_mof_rerelax)
# get adsorbate reference energy
h2o = mof_h2o[-3:]
h2o.calc = calc
E_h2o = h2o.get_potential_energy()
# compute adsorption energy
E_ads = E_combo - E_mof - E_h2o
print(f"Adsorption energy of H2O in WOBHEB_0.11_0: {E_ads:.3f} eV")Adsorption energy of H2O in WOBHEB_0.11_0: -0.539 eV
This adsorption energy closely matches that from DFT (–0.699 eV) [1]. The strong adsorption energy is a consequence of both H2O chemisorption and MOF deformation. We can decompose the adsorption energy into contributions from these two factors. Assuming rigid H2O molecules, we define and , respectively, as
describes host host–guest interactions for the MOF in the adsorbed state only. quantifies the magnitude of deformation between the MOF in the adsorbed state and the most energetically favorable empty MOF geometry determined from the workflow presented here. It can be shown that
For H2O adsorption in WOBHEB_0.11, we have
E_int = E_combo - E_mof_adsorbed_state - E_h2o
print(f"E_int: {E_int}")E_int: -0.8252324528776267
E_mof_deform = E_mof_adsorbed_state - E_mof_empty
print(f"E_mof_deform: {E_mof_deform}")E_mof_deform: 0.28575044860713206
E_ads = E_int + E_mof_deform
print(f"E_ads: {E_ads}")E_ads: -0.5394820042704946
is equivalent to when the MOF is assumed to be rigid. In this case, failure to consider adsorbate-induced deformation would result in an overestimation of the adsorption energy magnitude.
Acknowledgements & Authors¶
Logan Brabson and Sihoon Choi (Georgia Tech) and the OpenDAC project.
- Sriram, A., Choi, S., Yu, X., Brabson, L. M., Das, A., Ulissi, Z., Uyttendaele, M., Medford, A. J., & Sholl, D. S. (2024). The Open DAC 2023 Dataset and Challenges for Sorbent Discovery in Direct Air Capture. ACS Central Science, 10(5), 923–941. 10.1021/acscentsci.3c01629
- Queen, W. L., Hudson, M. R., Bloch, E. D., Mason, J. A., Gonzalez, M. I., Lee, J. S., Gygi, D., Howe, J. D., Lee, K., Darwish, T. A., James, M., Peterson, V. K., Teat, S. J., Smit, B., Neaton, J. B., Long, J. R., & Brown, C. M. (2014). Comprehensive study of carbon dioxide adsorption in the metal–organic frameworks M 2 (dobdc) (M = Mg, Mn, Fe, Co, Ni, Cu, Zn). Chem. Sci., 5(12), 4569–4581. 10.1039/c4sc02064b
- Yu, D., Yazaydin, A. O., Lane, J. R., Dietzel, P. D. C., & Snurr, R. Q. (2013). A combined experimental and quantum chemical study of CO2 adsorption in the metal–organic framework CPO-27 with different metals. Chemical Science, 4(9), 3544. 10.1039/c3sc51319j
- Alonso, G., Bahamon, D., Keshavarz, F., Giménez, X., Gamallo, P., & Sayós, R. (2018). Density Functional Theory-Based Adsorption Isotherms for Pure and Flue Gas Mixtures on Mg-MOF-74. Application in CO2 Capture Swing Adsorption Processes. The Journal of Physical Chemistry C, 122(7), 3945–3957. 10.1021/acs.jpcc.8b00938
- Witman, M., Ling, S., Jawahery, S., Boyd, P. G., Haranczyk, M., Slater, B., & Smit, B. (2017). The Influence of Intrinsic Framework Flexibility on Adsorption in Nanoporous Materials. Journal of the American Chemical Society, 139(15), 5547–5557. 10.1021/jacs.7b01688