This tutorial will walk you through a few examples of how you can use UMA. Each step is covered in more detail elsewhere in the documentation, but this is well suited to a ~1-2 hour tutorial session for researchers new to UMA but with some background in ASE and molecular simulations.
Before you start / installation¶
You need to get a HuggingFace account and request access to the UMA models.
You need a Huggingface account, request access to https://
Permissions: Read access to contents of all public gated repos you can access
Then, add the token as an environment variable (using huggingface-cli login:
# Enter token via huggingface-cli
! huggingface-cli loginor you can set the token via HF_TOKEN variable:
# Set token via env variable
import os
os.environ['HF_TOKEN'] = 'MYTOKEN'Installation process¶
It may be enough to use pip install fairchem-core. This gets you the latest version on PyPi (https://
Here we install some sub-packages. This can take 2-5 minutes to run.
! pip install fairchem-core fairchem-data-oc fairchem-applications-cattsunami x3dase# Check that packages are installed
!pip list | grep fairchemfairchem-applications-cattsunami 1.1.2.dev362+g4632f926a
fairchem-core 2.21.1.dev62+g4632f926a
fairchem-data-oc 1.0.3.dev362+g4632f926a
fairchem-data-omat 0.2.1.dev267+g4632f926a
import fairchem.core
fairchem.core.__version__'2.21.1.dev62+g4632f926a'Illustrative examples¶
These should just run, and are here to show some basic uses.
Spin gap energy - OMOL¶
This is the difference in energy between a triplet and single ground state for a CH2 radical. This downloads a ~1GB checkpoint the first time you run it.
We don’t set a device here, so we get a warning about using a CPU device. You can ignore that. If a CUDA environment is available, a GPU may be used to speed up the calculations.
from fairchem.core import FAIRChemCalculator, pretrained_mlip
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
from ase.build import molecule
# singlet CH2
singlet = molecule("CH2_s1A1d")
singlet.info.update({"spin": 1, "charge": 0})
singlet.calc = FAIRChemCalculator(predictor, task_name="omol")
# triplet CH2
triplet = molecule("CH2_s3B1d")
triplet.info.update({"spin": 3, "charge": 0})
triplet.calc = FAIRChemCalculator(predictor, task_name="omol")
print(triplet.get_potential_energy() - singlet.get_potential_energy())WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
W0807 05:47:46.005000 8234 site-packages/torch/_logging/_internal.py:1345] [0/0] Profiler record function <class 'torch.autograd.profiler.record_function'> will be ignored
W0807 05:47:53.806000 8234 site-packages/torch/_inductor/utils.py:1953] [3/0_1] Not enough SMs to use max_autotune_gemm mode
/home/runner/work/_tool/Python/3.12.13/x64/lib/python3.12/site-packages/torch/_inductor/lowering.py:2352: FutureWarning: `torch._prims_common.check` is deprecated and will be removed in the future. Please use `torch._check*` functions instead.
check(
WARNING:root:The UMA fast path (merge_mole + compile) is only available for fixed composition, task, charge, and spin. This is optimized for MD applications. Falling back to a less optimized version for subsequent evaluations. Reason: 'Spin differs: tensor([3]) vs tensor([1], device='cuda:0')'.
Use inference_settings='batch' for heterogeneous batched evaluations.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
-0.6104822014046931
Example of adsorbate relaxation - OC20¶
Here we just setup a Cu(100) slab with a CO on it and relax it.
We specify an explicit device in the predictor here, and avoid the warning.
from ase.build import add_adsorbate, fcc100, molecule
from ase.optimize import LBFGS
from fairchem.core import FAIRChemCalculator, pretrained_mlip
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(predictor, task_name="oc20")
# Set up your system as an ASE atoms object
slab = fcc100("Cu", (3, 3, 3), vacuum=8, periodic=True)
adsorbate = molecule("CO")
add_adsorbate(slab, adsorbate, 2.0, "bridge")
slab.calc = calc
# Set up LBFGS dynamics object
opt = LBFGS(slab)
opt.run(0.05, 100)
print(slab.get_potential_energy())WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
/home/runner/work/_tool/Python/3.12.13/x64/lib/python3.12/site-packages/torch/_inductor/lowering.py:2352: FutureWarning: `torch._prims_common.check` is deprecated and will be removed in the future. Please use `torch._check*` functions instead.
check(
Step Time Energy fmax
LBFGS: 0 05:51:11 -89.544141 11.402925
LBFGS: 1 05:51:12 -92.457701 6.589585
LBFGS: 2 05:51:12 -92.585267 7.565845
LBFGS: 3 05:51:12 -92.966559 3.751869
LBFGS: 4 05:51:12 -93.124915 3.558085
LBFGS: 5 05:51:12 -93.231281 2.245462
LBFGS: 6 05:51:12 -93.471494 1.133978
LBFGS: 7 05:51:12 -93.563004 0.996111
LBFGS: 8 05:51:12 -93.673188 0.698586
LBFGS: 9 05:51:13 -93.760223 0.492598
LBFGS: 10 05:51:13 -93.806521 0.365443
LBFGS: 11 05:51:13 -93.825451 0.344569
LBFGS: 12 05:51:13 -93.849869 0.486306
LBFGS: 13 05:51:13 -93.868569 0.424174
LBFGS: 14 05:51:13 -93.878644 0.154713
LBFGS: 15 05:51:13 -93.884597 0.169649
LBFGS: 16 05:51:13 -93.891121 0.207652
LBFGS: 17 05:51:13 -93.897926 0.250701
LBFGS: 18 05:51:13 -93.903971 0.173374
LBFGS: 19 05:51:13 -93.906880 0.053486
LBFGS: 20 05:51:14 -93.907327 0.042110
-93.9073274573666
Example bulk relaxation - OMAT¶
from ase.build import bulk
from ase.filters import FrechetCellFilter
from ase.optimize import FIRE
from fairchem.core import FAIRChemCalculator, pretrained_mlip
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(predictor, task_name="omat")
atoms = bulk("Fe")
atoms.calc = calc
opt = FIRE(FrechetCellFilter(atoms))
opt.run(0.05, 100)
print(atoms.get_stress()) # !!!! We get stress now!WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
Step Time Energy fmax
FIRE: 0 05:53:02 -8.263804 0.571333
FIRE: 1 05:53:03 -8.270171 0.139049
FIRE: 2 05:53:03 -8.259920 1.610407
FIRE: 3 05:53:04 -8.269784 0.262889
FIRE: 4 05:53:04 -8.269145 0.282856
FIRE: 5 05:53:05 -8.269291 0.268282
FIRE: 6 05:53:05 -8.269553 0.239071
FIRE: 7 05:53:05 -8.269875 0.195163
FIRE: 8 05:53:05 -8.270182 0.136499
FIRE: 9 05:53:06 -8.270394 0.063366
FIRE: 10 05:53:06 -8.270440 0.023308
[-2.01449794e-03 -2.01410064e-03 -2.01488310e-03 8.30762753e-09
-1.81644709e-09 -9.94237221e-10]
Molecular dynamics - OMOL¶
import matplotlib.pyplot as plt
from ase import units
from ase.build import molecule
from ase.io import Trajectory
from ase.md.langevin import Langevin
from fairchem.core import FAIRChemCalculator, pretrained_mlip
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(predictor, task_name="omol")
atoms = molecule("H2O")
atoms.info.update(charge=0, spin=1) # For omol
atoms.calc = calc
dyn = Langevin(
atoms,
timestep=0.1 * units.fs,
temperature_K=400,
friction=0.001 / units.fs,
)
trajectory = Trajectory("my_md.traj", "w", atoms)
dyn.attach(trajectory.write, interval=1)
dyn.run(steps=50)
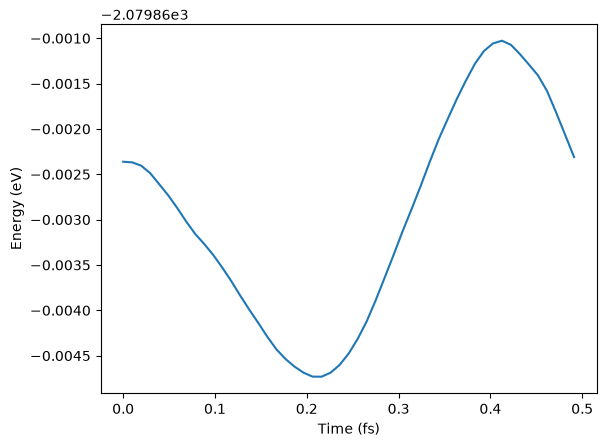
# See some results - not paper ready!
traj = Trajectory("my_md.traj")
plt.plot(
[i * 0.1 * units.fs for i in range(len(traj))],
[a.get_potential_energy() for a in traj],
)
plt.xlabel("Time (fs)")
plt.ylabel("Energy (eV)");WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
/home/runner/work/_tool/Python/3.12.13/x64/lib/python3.12/site-packages/ase/md/langevin.py:102: FutureWarning: The implementation of `fixcm=True` in `Langevin` does not strictly sample the correct NVT distributions. The deviations are typically small for large systems but can be more pronounced for small systems. Use `fixcm=False` together with `ase.constraints.FixCom`. `fixcm` is deprecated since ASE 3.28.0 and will be removed in a future release.
warnings.warn(msg, FutureWarning)
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
Catalyst Adsorption energies¶
The basic approach in computing an adsorption energy is to compute this energy difference:
dH = E_adslab - E_slab - E_adsWe use UMA for two of these energies E_adslab and E_slab. For E_ads We have to do something a little different. The OC20 task is not trained for molecules or molecular fragments. We use atomic energy reference energies instead. These are tabulated below.
The OC20 reference scheme is this reaction:
x CO + (x + y/2 - z) H2 + (z-x) H2O + w/2 N2 + * -> CxHyOzNw* For this example we have
-H2 + H2O + * -> O*. "O": -7.204 eVWhere "O": -7.204 is a constant.
To get the desired reaction energy we want we add the formation energy of water. We use either DFT or experimental values for this reaction energy.
1/2O2 + H2 -> H2OAlternatives to this approach are using DFT to estimate the energy of 1/2 O2, just make sure to use consistent settings with your task. You should not use OMOL for this.
from ase.build import add_adsorbate, fcc111
from ase.optimize import BFGS
from fairchem.core import FAIRChemCalculator, pretrained_mlip
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(predictor, task_name="oc20")WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
# reference energies from a linear combination of H2O/N2/CO/H2!
atomic_reference_energies = {
"H": -3.477,
"N": -8.083,
"O": -7.204,
"C": -7.282,
}
re1 = -3.03 # Water formation energy from experiment
slab = fcc111("Pt", size=(2, 2, 5), vacuum=20.0)
slab.pbc = True
adslab = slab.copy()
add_adsorbate(adslab, "O", height=1.2, position="fcc")
slab.calc = calc
opt = BFGS(slab)
print("Relaxing slab")
opt.run(fmax=0.05, steps=100)
slab_e = slab.get_potential_energy()
adslab.calc = calc
opt = BFGS(adslab)
print("\nRelaxing adslab")
opt.run(fmax=0.05, steps=100)
adslab_e = adslab.get_potential_energy()Relaxing slab
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
Step Time Energy fmax
BFGS: 0 05:53:22 -104.695194 0.709590
BFGS: 1 05:53:22 -104.753189 0.607749
BFGS: 2 05:53:22 -104.906055 0.369388
BFGS: 3 05:53:22 -104.938751 0.439760
BFGS: 4 05:53:22 -105.016145 0.464012
BFGS: 5 05:53:22 -105.076564 0.356073
WARNING:root:The UMA fast path (merge_mole + compile) is only available for fixed composition, task, charge, and spin. This is optimized for MD applications. Falling back to a less optimized version for subsequent evaluations. Reason: 'Compositions differ from merged model'.
Use inference_settings='batch' for heterogeneous batched evaluations.
BFGS: 6 05:53:22 -105.112620 0.189412
BFGS: 7 05:53:22 -105.126759 0.045357
Relaxing adslab
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
Step Time Energy fmax
BFGS: 0 05:53:28 -110.048788 1.756661
BFGS: 1 05:53:28 -110.230718 0.986315
BFGS: 2 05:53:28 -110.379375 0.731556
BFGS: 3 05:53:29 -110.430299 0.807226
BFGS: 4 05:53:29 -110.543733 0.691539
BFGS: 5 05:53:29 -110.617504 0.492725
BFGS: 6 05:53:29 -110.673479 0.667091
BFGS: 7 05:53:30 -110.721707 0.710397
BFGS: 8 05:53:30 -110.756917 0.443563
BFGS: 9 05:53:30 -110.769297 0.214378
BFGS: 10 05:53:30 -110.772451 0.091669
BFGS: 11 05:53:30 -110.772964 0.062458
BFGS: 12 05:53:31 -110.773257 0.046191
Now we compute the adsorption energy.
# Energy for ((H2O-H2) + * -> *O) + (H2 + 1/2O2 -> H2O) leads to 1/2O2 + * -> *O!
adslab_e - slab_e - atomic_reference_energies["O"] + re1-1.472498324072013How did we do? We need a reference point. In the paper below, there is an atomic adsorption energy for O on Pt(111) of about -4.264 eV. This is for the reaction O + * -> O*. To convert this to the dissociative adsorption energy, we have to add the reaction:
1/2 O2 -> O D = 2.58 eV (expt)to get a comparable energy of about -1.68 eV. There is about ~0.2 eV difference (we predicted -1.47 eV above, and the reference comparison is -1.68 eV) to account for. The biggest difference is likely due to the differences in exchange-correlation functional. The reference data used the PBE functional, and eSCN was trained on RPBE data. To additional places where there are differences include:
Difference in lattice constant
The reference energy used for the experiment references. These can differ by up to 0.5 eV from comparable DFT calculations.
How many layers are relaxed in the calculation
Some of these differences tend to be systematic, and you can calibrate and correct these, especially if you can augment these with your own DFT calculations.
It is always a good idea to visualize the geometries to make sure they look reasonable.
import matplotlib.pyplot as plt
from ase.visualize.plot import plot_atoms
fig, axs = plt.subplots(1, 2)
plot_atoms(slab, axs[0])
plot_atoms(slab, axs[1], rotation=("-90x"))
axs[0].set_axis_off()
axs[1].set_axis_off()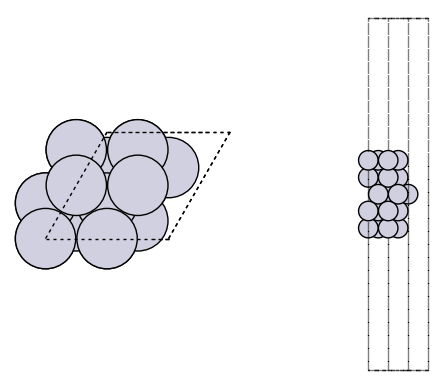
fig, axs = plt.subplots(1, 2)
plot_atoms(adslab, axs[0])
plot_atoms(adslab, axs[1], rotation=("-90x"))
axs[0].set_axis_off()
axs[1].set_axis_off()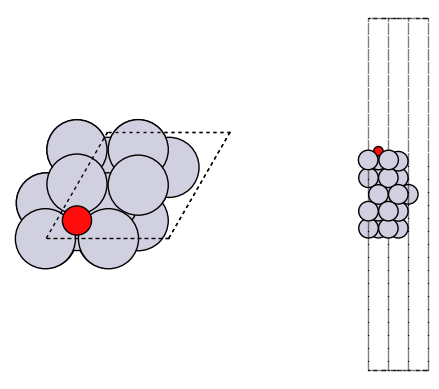
Molecular vibrations¶
from ase import Atoms
from ase.optimize import BFGS
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(predictor, task_name="omol")
from ase.vibrations import Vibrations
n2 = Atoms("N2", [(0, 0, 0), (0, 0, 1.1)])
n2.info.update({"spin": 1, "charge": 0})
n2.calc = calc
BFGS(n2).run(fmax=0.01)WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
Step Time Energy fmax
BFGS: 0 05:53:40 -2981.069021 1.646100
BFGS: 1 05:53:40 -2980.962520 6.604125
BFGS: 2 05:53:40 -2981.077760 0.200669
BFGS: 3 05:53:40 -2981.077885 0.023347
BFGS: 4 05:53:40 -2981.077887 0.000100
np.True_vib = Vibrations(n2)
vib.run()
vib.summary()---------------------
# meV cm^-1
---------------------
0 0.0i 0.0i
1 0.0i 0.0i
2 0.0i 0.0i
3 2.0 16.1
4 2.0 16.1
5 309.6 2496.7
---------------------
Zero-point energy: 0.157 eV
Bulk alloy phase behavior¶
Adapted from https://
We manually compute the formation energy of pure compounds and some alloy compositions to assess stability.
from ase.atoms import Atom, Atoms
from ase.filters import FrechetCellFilter
from ase.optimize import FIRE
from fairchem.core import FAIRChemCalculator, pretrained_mlip
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")
cu = Atoms(
[Atom("Cu", [0.000, 0.000, 0.000])],
cell=[[1.818, 0.000, 1.818], [1.818, 1.818, 0.000], [0.000, 1.818, 1.818]],
pbc=True,
)
cu.calc = FAIRChemCalculator(predictor, task_name="omat")
opt = FIRE(FrechetCellFilter(cu))
opt.run(0.05, 100)
cu.get_potential_energy()WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
Step Time Energy fmax
FIRE: 0 05:53:47 -3.748689 0.185703
FIRE: 1 05:53:47 -3.749557 0.126066
FIRE: 2 05:53:47 -3.750259 0.023407
-3.750259493460482pd = Atoms(
[Atom("Pd", [0.000, 0.000, 0.000])],
cell=[[1.978, 0.000, 1.978], [1.978, 1.978, 0.000], [0.000, 1.978, 1.978]],
pbc=True,
)
pd.calc = FAIRChemCalculator(predictor, task_name="omat")
opt = FIRE(FrechetCellFilter(pd))
opt.run(0.05, 100)
pd.get_potential_energy()WARNING:root:The UMA fast path (merge_mole + compile) is only available for fixed composition, task, charge, and spin. This is optimized for MD applications. Falling back to a less optimized version for subsequent evaluations. Reason: 'Compositions differ from merged model'.
Use inference_settings='batch' for heterogeneous batched evaluations.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
Step Time Energy fmax
FIRE: 0 05:53:53 -5.208746 0.206138
FIRE: 1 05:53:54 -5.209731 0.111874
FIRE: 2 05:53:54 -5.210079 0.040758
-5.210078715135518Alloy formation energies¶
cupd1 = Atoms(
[Atom("Cu", [0.000, 0.000, 0.000]), Atom("Pd", [-1.652, 0.000, 2.039])],
cell=[[0.000, -2.039, 2.039], [0.000, 2.039, 2.039], [-3.303, 0.000, 0.000]],
pbc=True,
) # Note pbc=True is important, it is not the default and OMAT
cupd1.calc = FAIRChemCalculator(predictor, task_name="omat")
opt = FIRE(FrechetCellFilter(cupd1))
opt.run(0.05, 100)
cupd1.get_potential_energy() Step Time Energy fmax
FIRE: 0 05:53:54 -9.200180 0.149828
FIRE: 1 05:53:55 -9.200405 0.137150
FIRE: 2 05:53:55 -9.200767 0.114193
FIRE: 3 05:53:56 -9.201159 0.085832
FIRE: 4 05:53:56 -9.201547 0.064592
FIRE: 5 05:53:56 -9.201993 0.080829
FIRE: 6 05:53:57 -9.202580 0.083421
FIRE: 7 05:53:57 -9.203317 0.072357
FIRE: 8 05:53:58 -9.204221 0.065461
FIRE: 9 05:53:58 -9.205216 0.090108
FIRE: 10 05:53:58 -9.206354 0.104151
FIRE: 11 05:53:59 -9.207779 0.094693
FIRE: 12 05:53:59 -9.209430 0.060872
FIRE: 13 05:54:00 -9.211120 0.055550
FIRE: 14 05:54:01 -9.212895 0.038677
-9.212894721737795cupd2 = Atoms(
[
Atom("Cu", [-0.049, 0.049, 0.049]),
Atom("Cu", [-11.170, 11.170, 11.170]),
Atom("Pd", [-7.415, 7.415, 7.415]),
Atom("Pd", [-3.804, 3.804, 3.804]),
],
cell=[[-5.629, 3.701, 5.629], [-3.701, 5.629, 5.629], [-5.629, 5.629, 3.701]],
pbc=True,
)
cupd2.calc = FAIRChemCalculator(predictor, task_name="omat")
opt = FIRE(FrechetCellFilter(cupd2))
opt.run(0.05, 100)
cupd2.get_potential_energy() Step Time Energy fmax
FIRE: 0 05:54:01 -18.132613 0.170385
FIRE: 1 05:54:02 -18.133448 0.153581
FIRE: 2 05:54:02 -18.134793 0.121402
FIRE: 3 05:54:02 -18.136119 0.076687
FIRE: 4 05:54:03 -18.136904 0.023653
-18.13690361998487# Delta Hf cupd-1 = -0.11 eV/atom
hf1 = (
cupd1.get_potential_energy() - cu.get_potential_energy() - pd.get_potential_energy()
)
hf1-0.25255651314179506# DFT: Delta Hf cupd-2 = -0.04 eV/atom
hf2 = (
cupd2.get_potential_energy()
- 2 * cu.get_potential_energy()
- 2 * pd.get_potential_energy()
)
hf2-0.21622720279287044hf1 - hf2, (-0.11 - -0.04)(-0.036329310348924615, -0.07)These indicate that cupd-1 and cupd-2 are both more stable than phase separated Cu and Pd, and that cupd-1 is more stable than cupd-2. The absolute formation energies differ from the DFT references, but the relative differences are quite close. The absolute differences could be due to DFT parameter choices (XC, psp, etc.).
Phonon calculation¶
This takes 4-10 minutes. Adapted from https://
Phonons have applications in computing the stability and free energy of solids. See:
https://
www .sciencedirect .com /science /article /pii /S1359646215003127 https://
iopscience .iop .org /book /mono /978 -0 -7503 -2572 -1 /chapter /bk978 -0 -7503 -2572 -1ch1
from ase.build import bulk
from ase.phonons import Phonons
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(predictor, task_name="omat")
# Setup crystal
atoms = bulk("Al", "fcc", a=4.05)
# Phonon calculator
N = 7
ph = Phonons(atoms, calc, supercell=(N, N, N), delta=0.05)
ph.run()
# Read forces and assemble the dynamical matrix
ph.read(acoustic=True)
ph.clean()
path = atoms.cell.bandpath("GXULGK", npoints=100)
bs = ph.get_band_structure(path)
dos = ph.get_dos(kpts=(20, 20, 20)).sample_grid(npts=100, width=1e-3)WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
WARNING, 2 imaginary frequencies at q = ( 0.00, 0.00, 0.00) ; (omega_q = 8.335e-09*i)
WARNING, 1 imaginary frequencies at q = ( 0.02, 0.02, 0.02) ; (omega_q = 1.432e-02*i)
WARNING, 2 imaginary frequencies at q = ( 0.00, 0.00, 0.00) ; (omega_q = 8.335e-09*i)
WARNING, 1 imaginary frequencies at q = ( 0.01, 0.01, 0.03) ; (omega_q = 1.659e-02*i)
WARNING, 1 imaginary frequencies at q = ( 0.03, 0.03, 0.05) ; (omega_q = 2.249e-02*i)
WARNING, 1 imaginary frequencies at q = ( 0.04, 0.04, 0.08) ; (omega_q = 2.057e-02*i)
WARNING, 1 imaginary frequencies at q = (-0.08, -0.03, -0.03) ; (omega_q = 1.618e-02*i)
WARNING, 1 imaginary frequencies at q = (-0.03, -0.08, -0.03) ; (omega_q = 1.618e-02*i)
WARNING, 1 imaginary frequencies at q = (-0.03, -0.03, -0.08) ; (omega_q = 1.619e-02*i)
WARNING, 1 imaginary frequencies at q = (-0.03, -0.03, -0.03) ; (omega_q = 1.444e-02*i)
WARNING, 1 imaginary frequencies at q = ( 0.03, 0.03, 0.03) ; (omega_q = 1.444e-02*i)
WARNING, 1 imaginary frequencies at q = ( 0.03, 0.03, 0.07) ; (omega_q = 1.619e-02*i)
WARNING, 1 imaginary frequencies at q = ( 0.03, 0.07, 0.03) ; (omega_q = 1.618e-02*i)
WARNING, 1 imaginary frequencies at q = ( 0.07, 0.03, 0.03) ; (omega_q = 1.618e-02*i)
# Plot the band structure and DOS:
import matplotlib.pyplot as plt # noqa
fig = plt.figure(figsize=(7, 4))
ax = fig.add_axes([0.12, 0.07, 0.67, 0.85])
emax = 0.04
bs.plot(ax=ax, emin=0.0, emax=emax)
dosax = fig.add_axes([0.8, 0.07, 0.17, 0.85])
dosax.fill_between(
dos.get_weights(),
dos.get_energies(),
y2=0,
color="grey",
edgecolor="k",
lw=1,
)
dosax.set_ylim(0, emax)
dosax.set_yticks([])
dosax.set_xticks([])
dosax.set_xlabel("DOS", fontsize=18);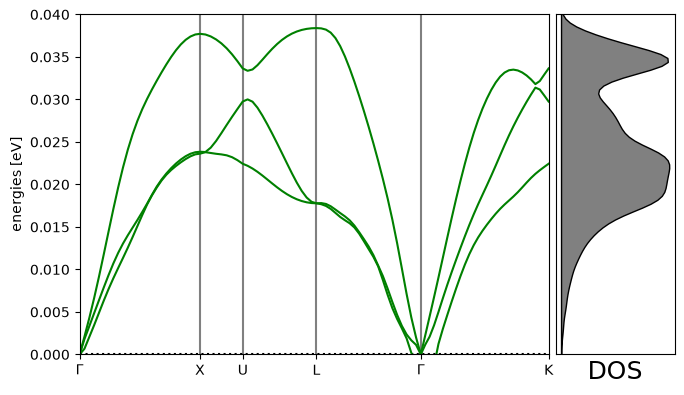
Transition States (NEBs)¶
Nudged elastic band calculations are among the most costly calculations we do. UMA makes these quicker!
We explore diffusion of an O adatom from an hcp to an fcc site on Pt(111).
Initial state¶
from ase.build import add_adsorbate, fcc111, molecule
from ase.optimize import LBFGS
from fairchem.core import FAIRChemCalculator, pretrained_mlip
predictor = pretrained_mlip.get_predict_unit("uma-s-1p1")
calc = FAIRChemCalculator(predictor, task_name="oc20")
# Set up your system as an ASE atoms object
initial = fcc111("Pt", (3, 3, 3), vacuum=8, periodic=True)
adsorbate = molecule("O")
add_adsorbate(initial, adsorbate, 2.0, "fcc")
initial.calc = calc
# Set up LBFGS dynamics object
opt = LBFGS(initial)
opt.run(0.05, 100)
print(initial.get_potential_energy())WARNING:root:device was not explicitly set, using device='cuda'.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:If 'dataset_list' is provided in the config, the code assumes that each dataset maps to itself. Please use 'dataset_mapping' as 'dataset_list' is deprecated and will be removed in the future.
WARNING:root:Model is being compiled this might take a while for the first time
Step Time Energy fmax
LBFGS: 0 05:54:19 -141.309108 3.517762
LBFGS: 1 05:54:19 -141.702377 3.524942
LBFGS: 2 05:54:19 -142.967947 2.974642
LBFGS: 3 05:54:19 -143.672289 0.963277
LBFGS: 4 05:54:19 -143.776589 1.269399
LBFGS: 5 05:54:20 -143.848508 0.873114
LBFGS: 6 05:54:20 -143.923349 0.164695
LBFGS: 7 05:54:20 -143.926599 0.153171
LBFGS: 8 05:54:20 -143.933658 0.126658
LBFGS: 9 05:54:20 -143.937971 0.116472
LBFGS: 10 05:54:20 -143.941407 0.073791
LBFGS: 11 05:54:20 -143.942885 0.083216
LBFGS: 12 05:54:20 -143.944361 0.084958
LBFGS: 13 05:54:20 -143.945986 0.065907
LBFGS: 14 05:54:21 -143.947507 0.037425
-143.9475067667952
Final state¶
# Set up your system as an ASE atoms object
final = fcc111("Pt", (3, 3, 3), vacuum=8, periodic=True)
adsorbate = molecule("O")
add_adsorbate(final, adsorbate, 2.0, "hcp")
final.calc = FAIRChemCalculator(predictor, task_name="oc20")
# Set up LBFGS dynamics object
opt = LBFGS(final)
opt.run(0.05, 100)
print(final.get_potential_energy()) Step Time Energy fmax
LBFGS: 0 05:54:21 -141.268799 3.362366
LBFGS: 1 05:54:21 -141.650621 3.342133
LBFGS: 2 05:54:21 -142.883883 2.576831
LBFGS: 3 05:54:21 -143.406004 1.208926
LBFGS: 4 05:54:21 -143.469218 0.959532
LBFGS: 5 05:54:21 -143.589296 0.127614
LBFGS: 6 05:54:22 -143.593122 0.112181
LBFGS: 7 05:54:22 -143.595709 0.091211
LBFGS: 8 05:54:22 -143.597250 0.071136
LBFGS: 9 05:54:22 -143.598559 0.047106
-143.59855861951266
Setup and relax the band¶
from ase.mep import NEB
images = [initial]
for i in range(3):
image = initial.copy()
image.calc = FAIRChemCalculator(predictor, task_name="oc20")
images.append(image)
images.append(final)
neb = NEB(images)
neb.interpolate()
opt = LBFGS(neb, trajectory="neb.traj")
opt.run(0.05, 100)/home/runner/work/_tool/Python/3.12.13/x64/lib/python3.12/site-packages/ase/mep/neb.py:329: UserWarning: The default method has changed from 'aseneb' to 'improvedtangent'. The 'aseneb' method is an unpublished, custom implementation that is not recommended as it frequently results in very poor bands. Please explicitly set method='improvedtangent' to silence this warning, or set method='aseneb' if you strictly require the old behavior (results may vary). See: https://gitlab.com/ase/ase/-/merge_requests/3952
warnings.warn(
Step Time Energy fmax
LBFGS: 0 05:54:22 -143.171396 3.064781
LBFGS: 1 05:54:23 -143.340627 1.486502
LBFGS: 2 05:54:23 -143.393669 0.434151
LBFGS: 3 05:54:23 -143.405931 0.453322
LBFGS: 4 05:54:24 -143.426327 0.462209
LBFGS: 5 05:54:24 -143.442347 0.346508
LBFGS: 6 05:54:25 -143.452654 0.180226
LBFGS: 7 05:54:25 -143.457226 0.151728
LBFGS: 8 05:54:25 -143.459749 0.175265
LBFGS: 9 05:54:26 -143.461191 0.188577
LBFGS: 10 05:54:26 -143.462572 0.176422
LBFGS: 11 05:54:27 -143.463357 0.094811
LBFGS: 12 05:54:27 -143.463786 0.090829
LBFGS: 13 05:54:27 -143.464269 0.101391
LBFGS: 14 05:54:28 -143.464863 0.128216
LBFGS: 15 05:54:28 -143.465402 0.087880
LBFGS: 16 05:54:28 -143.465722 0.047132
np.True_from ase.mep import NEBTools
NEBTools(neb.images).plot_band();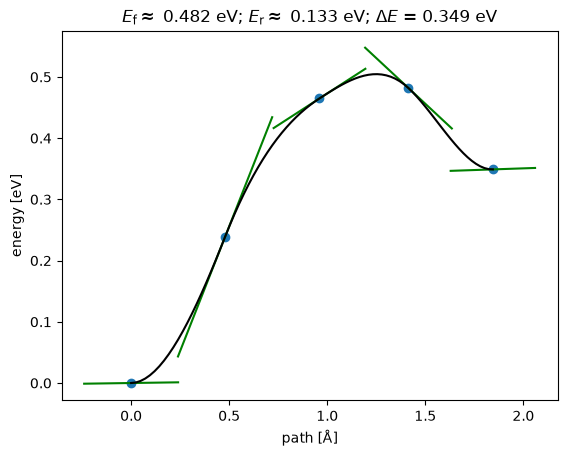
This could be a good initial guess to initialize an NEB in DFT.
Ideas for things you can do with UMA¶
Advanced applications¶
These take a while to run.
AdsorbML¶
It is so cheap to run these calculations that we can screen a broad range of adsorbate sites and rank them in stability. The AdsorbML approach automates this. This takes quite a while to run here, and we don’t do it in the workshop.
Expert adsorption energies¶
This tutorial reproduces Fig 6b from the following paper: Zhou, Jing, et al. “Enhanced Catalytic Activity of Bimetallic Ordered Catalysts for Nitrogen Reduction Reaction by Perturbation of Scaling Relations.” ACS Catalysis 134 (2023): 2190-2201 (Zhou et al. (2023)).
This takes up to an hour with a GPU, and much longer with a CPU.
CatTsunami¶
The CatTsunami tutorial is an example of enumerating initial and final states, and computing reaction paths between them with UMA.
Acknowledgments¶
This tutorial was originally compiled by John Kitchin (CMU) for the NAM29 catalysis tutorial session, using a variety of resources from the FAIR chemistry repository.
- Musielewicz, J., Wang, X., Tian, T., & Ulissi, Z. (2022). FINETUNA: fine-tuning accelerated molecular simulations. Machine Learning: Science and Technology, 3(3), 03LT01. 10.1088/2632-2153/ac8fe0
- Wang, X., Musielewicz, J., Tran, R., Kumar Ethirajan, S., Fu, X., Mera, H., Kitchin, J. R., Kurchin, R. C., & Ulissi, Z. W. (2024). Generalization of graph-based active learning relaxation strategies across materials. Machine Learning: Science and Technology, 5(2), 025018. 10.1088/2632-2153/ad37f0
- Wander, B., Musielewicz, J., Cheula, R., & Kitchin, J. R. (2025). Accessing Numerical Energy Hessians with Graph Neural Network Potentials and Their Application in Heterogeneous Catalysis. The Journal of Physical Chemistry C, 129(7), 3510–3521. 10.1021/acs.jpcc.4c07477
- Zhou, J., Chen, X., Guo, M., Hu, W., Huang, B., & Yuan, D. (2023). Enhanced Catalytic Activity of Bimetallic Ordered Catalysts for Nitrogen Reduction Reaction by Perturbation of Scaling Relations. ACS Catalysis, 13(4), 2190–2201. 10.1021/acscatal.2c05877