# MIT License
#
#@title Copyright (c) 2021 CCAI Community Authors { display-mode: "form" }
#
# Permission is hereby granted, free of charge, to any person obtaining a
# copy of this software and associated documentation files (the "Software"),
# to deal in the Software without restriction, including without limitation
# the rights to use, copy, modify, merge, publish, distribute, sublicense,
# and/or sell copies of the Software, and to permit persons to whom the
# Software is furnished to do so, subject to the following conditions:
#
# The above copyright notice and this permission notice shall be included in
# all copies or substantial portions of the Software.
#
# THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR
# IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY,
# FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL
# THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER
# LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING
# FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER
# DEALINGS IN THE SOFTWARE.
Open Catalyst Project Tutorial Notebook#
Author(s):
Muhammed Shuaibi, CMU, mshuaibi@andrew.cmu.edu
Abhishek Das, FAIR, abhshkdz@fb.com
Adeesh Kolluru, CMU, akolluru@andrew.cmu.edu
Brandon Wood, NERSC, bwood@lbl.gov
Janice Lan, FAIR, janlan@fb.com
Anuroop Sriram, FAIR, anuroops@fb.com
Zachary Ulissi, CMU, zulissi@andrew.cmu.edu
Larry Zitnick, FAIR, zitnick@fb.com
FAIR - Facebook AI Research
CMU - Carnegie Mellon University
NERSC - National Energy Research Scientific Computing Center
Background #
The discovery of efficient and economic catalysts (materials) are needed to enable the widespread use of renewable energy technologies. A common approach in discovering high performance catalysts is using molecular simulations. Specifically, each simulation models the interaction of a catalyst surface with molecules that are commonly seen in electrochemical reactions. By predicting these interactions accurately, the catalyst’s impact on the overall rate of a chemical reaction may be estimated.
An important quantity in screening catalysts is their adsorption energy for the molecules, referred to as `adsorbates’, involved in the reaction of interest. The adsorption energy may be found by simulating the interaction of the adsorbate molecule on the surface of the catalyst to find their resting or relaxed energy, i.e., how tightly the adsorbate binds to the catalyst’s surface (visualized below). The rate of the chemical reaction, a value of high practical importance, is then commonly approximated using simple functions of the adsorption energy. The goal of this tutorial specifically and the project overall is to encourage research and benchmark progress towards training ML models to approximate this relaxation.
Specifically, during the course of a relaxation, given an initial set of atoms and their positions, the task is to iteratively estimate atomic forces and update atomic positions until a relaxed state is reached. The energy corresponding to the relaxed state is the structure’s ‘relaxed energy’.
As part of the Open Catalyst Project (OCP), we identify three key tasks ML models need to perform well on in order to effectively approximate DFT –
Given an Initial Structure, predict the Relaxed Energy of the relaxed strucutre (IS2RE),
Given an Initial Structure, predict the Relaxed Structure (IS2RS),
Given any Structure, predict the structure Energy and per-atom Forces (S2EF).

Objective #
This notebook serves as a tutorial for interacting with the Open Catalyst Project.
By the end of this tutorial, users will have gained:
Intuition to the dataset and it’s properties
Knowledge of the various OCP tasks: IS2RE, IS2RS, S2EF
Steps to train, validate, and predict a model on the various tasks
A walkthrough on creating your own model
(Optional) Creating your own dataset for other molecular/catalyst applications
(Optional) Using pretrained models directly with an [ASE](https://wiki.fysik.dtu.dk/ase/#:~:text=The%20Atomic%20Simulation%20Environment%20(ASE,under%20the%20GNU%20LGPL%20license.)-style calculator.
Climate Impact#
Scalable and cost-effective solutions to renewable energy storage are essential to addressing the world’s rising energy needs while reducing climate change. As illustrated in the figure below, as we increase our reliance on renewable energy sources such as wind and solar, which produce intermittent power, storage is needed to transfer power from times of peak generation to peak demand. This may require the storage of power for hours, days, or months. One solution that offers the potential of scaling to nation-sized grids is the conversion of renewable energy to other fuels, such as hydrogen. To be widely adopted, this process requires cost-effective solutions to running chemical reactions.
An open challenge is finding low-cost catalysts to drive these reactions at high rates. Through the use of quantum mechanical simulations (Density Functional Theory, DFT), new catalyst structures can be tested and evaluated. Unfortunately, the high computational cost of these simulations limits the number of structures that may be tested. The use of AI or machine learning may provide a method to efficiently approximate these calculations; reducing the time required from 24} hours to a second. This capability would transform the search for new catalysts from the present day practice of evaluating O(1,000) of handpicked candidates to the brute force search over millions or even billions of candidates.
As part of OCP, we publicly released the world’s largest quantum mechanical simulation dataset – OC20 – in the Fall of 2020 along with a suite of baselines and evaluation metrics. The creation of the dataset required over 70 million hours of compute. This dataset enables the exploration of techniques that will generalize across different catalyst materials and adsorbates. If successful, models trained on the dataset could enable the computational testing of millions of catalyst materials for a wide variety of chemical reactions. However, techniques that achieve the accuracies required** for practical impact are still beyond reach and remain an open area for research, thus encouraging research in this important area to help in meeting the world’s energy needs in the decades ahead.
** The computational catalysis community often aims for an adsorption energy MAE of 0.1-0.2 eV for practical relevance.

Target Audience#
This tutorial is designed for those interested in application of ML towards climate change. More specifically, those interested in material/catalyst discovery and Graph Nueral Networks (GNNs) will find lots of benefit here. Little to no domain chemistry knowledge is necessary as it will be covered in the tutorial. Experience with GNNs is a plus but not required.
We have designed this notebook in a manner to get the ML communnity up to speed as far as background knowledge is concerned, and the catalysis community to better understand how to use the OCP’s state-of-the-art models in their everyday workflows.
Background & Prerequisites#
Basic experience training ML models. Familiarity with PyTorch. Familiarity with Pytorch-Geometric could be helpful for development, but not required. No background in chemistry is assumed.
For those looking to apply our pretrained models on their datasets, familiarity with the [Atomic Simulation Environment](https://wiki.fysik.dtu.dk/ase/#:~:text=The%20Atomic%20Simulation%20Environment%20(ASE,under%20the%20GNU%20LGPL%20license.) is useful.
Background References#
To gain an even better understanding of the Open Catalyst Project and the problems it seeks to address, we strongly recommend the following resources:
To learn more about electrocatalysis, see our white paper.
To learn about the OC20 dataset and the associated tasks, please see the OC20 dataset paper.
Software Requirements#
See installation for installation instructions!
import torch
torch.cuda.is_available()
False
Dataset Overview#
The Open Catalyst 2020 Dataset (OC20) will be used throughout this tutorial. More details can be found here and the corresponding paper. Data is stored in PyTorch Geometric Data objects and stored in LMDB files. For each task we include several sized training splits. Validation/Test splits are broken into several subsplits: In Domain (ID), Out of Domain Adsorbate (OOD-Ads), Out of Domain Catalyast (OOD-Cat) and Out of Domain Adsorbate and Catalyst (OOD-Both). Split sizes are summarized below:
Train
S2EF - 200k, 2M, 20M, 134M(All)
IS2RE/IS2RS - 10k, 100k, 460k(All)
Val/Test
S2EF - ~1M across all subsplits
IS2RE/IS2RS - ~25k across all splits
Tutorial Use#
For the sake of this tutorial we provide much smaller splits (100 train, 20 val for all tasks) to allow users to easily store, train, and predict across the various tasks. Please refer here for details on how to download the full datasets for general use.

Data Download [~1min] #
FOR TUTORIAL USE ONLY
%%bash
mkdir data
cd data
wget -q http://dl.fbaipublicfiles.com/opencatalystproject/data/tutorial_data.tar.gz -O tutorial_data.tar.gz
tar -xzvf tutorial_data.tar.gz
rm tutorial_data.tar.gz
mkdir: cannot create directory ‘data’: File exists
./
./is2re/
./is2re/train_100/
./is2re/train_100/data.lmdb
./is2re/train_100/data.lmdb-lock
./is2re/val_20/
./is2re/val_20/data.lmdb
./is2re/val_20/data.lmdb-lock
./s2ef/
./s2ef/train_100/
./s2ef/train_100/data.lmdb
./s2ef/train_100/data.lmdb-lock
./s2ef/val_20/
./s2ef/val_20/data.lmdb
./s2ef/val_20/data.lmdb-lock
Data Visualization #
import matplotlib
matplotlib.use('Agg')
import os
import numpy as np
import matplotlib.pyplot as plt
%matplotlib inline
params = {
'axes.labelsize': 14,
'font.size': 14,
'font.family': ' DejaVu Sans',
'legend.fontsize': 20,
'xtick.labelsize': 20,
'ytick.labelsize': 20,
'axes.labelsize': 25,
'axes.titlesize': 25,
'text.usetex': False,
'figure.figsize': [12, 12]
}
matplotlib.rcParams.update(params)
import ase.io
from ase.io.trajectory import Trajectory
from ase.io import extxyz
from ase.calculators.emt import EMT
from ase.build import fcc100, add_adsorbate, molecule
from ase.constraints import FixAtoms
from ase.optimize import LBFGS
from ase.visualize.plot import plot_atoms
from ase import Atoms
from IPython.display import Image
Understanding the data#
We use the Atomic Simulation Environment (ASE) library to interact with our data. This notebook will provide you with some intuition on how atomic data is generated, how the data is structured, how to visualize the data, and the specific properties that are passed on to our models.
Generating sample data#
The OC20 dataset was generated using density functional theory (DFT), a quantum chemistry method for modeling atomistic environments. For more details, please see our dataset paper. In this notebook, we generate sample data in the same format as the OC20 dataset; however, we use a faster method that is less accurate called effective-medium theory (EMT) because our DFT calculations are too computationally expensive to run here. EMT is great for demonstration purposes but not accurate enough for our actual catalysis applications. Below is a structural relaxation of a catalyst system, a propane (C3H8) adsorbate on a copper (Cu) surface. Throughout this tutorial a surface may be referred to as a slab and the combination of an adsorbate and a surface as an adslab.
Structural relaxations#
A structural relaxation or structure optimization is the process of iteratively updating atom positions to find the atom positions that minimize the energy of the structure. Standard optimization methods are used in structural relaxations — below we use the Limited-Memory Broyden–Fletcher–Goldfarb–Shanno (LBFGS) algorithm. The step number, time, energy, and force max are printed at each optimization step. Each step is considered one example because it provides all the information we need to train models for the S2EF task and the entire set of steps is referred to as a trajectory. Visualizing intermediate structures or viewing the entire trajectory can be illuminating to understand what is physically happening and to look for problems in the simulation, especially when we run ML-driven relaxations. Common problems one may look out for - atoms excessively overlapping/colliding with each other and atoms flying off into random directions.
###DATA GENERATION - FEEL FREE TO SKIP###
# This cell sets up and runs a structural relaxation
# of a propane (C3H8) adsorbate on a copper (Cu) surface
adslab = fcc100("Cu", size=(3, 3, 3))
adsorbate = molecule("C3H8")
add_adsorbate(adslab, adsorbate, 3, offset=(1, 1)) # adslab = adsorbate + slab
# tag all slab atoms below surface as 0, surface as 1, adsorbate as 2
tags = np.zeros(len(adslab))
tags[18:27] = 1
tags[27:] = 2
adslab.set_tags(tags)
# Fixed atoms are prevented from moving during a structure relaxation.
# We fix all slab atoms beneath the surface.
cons= FixAtoms(indices=[atom.index for atom in adslab if (atom.tag == 0)])
adslab.set_constraint(cons)
adslab.center(vacuum=13.0, axis=2)
adslab.set_pbc(True)
adslab.set_calculator(EMT())
os.makedirs('data', exist_ok=True)
# Define structure optimizer - LBFGS. Run for 100 steps,
# or if the max force on all atoms (fmax) is below 0 ev/A.
# fmax is typically set to 0.01-0.05 eV/A,
# for this demo however we run for the full 100 steps.
dyn = LBFGS(adslab, trajectory="data/toy_c3h8_relax.traj")
dyn.run(fmax=0, steps=100)
traj = ase.io.read("data/toy_c3h8_relax.traj", ":")
# convert traj format to extxyz format (used by OC20 dataset)
columns = (['symbols','positions', 'move_mask', 'tags', 'forces'])
with open('data/toy_c3h8_relax.extxyz','w') as f:
extxyz.write_xyz(f, traj, columns=columns)
Step Time Energy fmax
LBFGS: 0 04:43:29 15.804700 6.776430
LBFGS: 1 04:43:29 12.190607 4.323222
LBFGS: 2 04:43:29 10.240169 2.265527
LBFGS: 3 04:43:29 9.779223 0.937247
LBFGS: 4 04:43:29 9.671525 0.770173
LBFGS: 5 04:43:29 9.574461 0.663540
LBFGS: 6 04:43:29 9.537502 0.571800
LBFGS: 7 04:43:29 9.516673 0.446620
LBFGS: 8 04:43:29 9.481330 0.461143
LBFGS: 9 04:43:29 9.462255 0.293081
LBFGS: 10 04:43:29 9.448937 0.249010
LBFGS: 11 04:43:29 9.433813 0.237051
LBFGS: 12 04:43:29 9.418884 0.260245
LBFGS: 13 04:43:29 9.409649 0.253162
LBFGS: 14 04:43:29 9.404838 0.162398
LBFGS: 15 04:43:29 9.401753 0.182298
LBFGS: 16 04:43:29 9.397314 0.259163
LBFGS: 17 04:43:29 9.387947 0.345022
/tmp/ipykernel_3934/747130225.py:23: FutureWarning: Please use atoms.calc = calc
adslab.set_calculator(EMT())
LBFGS: 18 04:43:29 9.370825 0.407041
LBFGS: 19 04:43:29 9.342222 0.433340
LBFGS: 20 04:43:29 9.286822 0.500200
LBFGS: 21 04:43:29 9.249910 0.524052
LBFGS: 22 04:43:29 9.187179 0.511994
LBFGS: 23 04:43:29 9.124811 0.571796
LBFGS: 24 04:43:29 9.066185 0.540934
LBFGS: 25 04:43:30 9.000116 1.079833
LBFGS: 26 04:43:30 8.893632 0.752759
LBFGS: 27 04:43:30 8.845939 0.332051
LBFGS: 28 04:43:30 8.815173 0.251242
LBFGS: 29 04:43:30 8.808721 0.214337
LBFGS: 30 04:43:30 8.794643 0.154611
LBFGS: 31 04:43:30 8.789162 0.201404
LBFGS: 32 04:43:30 8.782320 0.175517
LBFGS: 33 04:43:30 8.780394 0.103718
LBFGS: 34 04:43:30 8.778410 0.107611
LBFGS: 35 04:43:30 8.775079 0.179747
LBFGS: 36 04:43:30 8.766987 0.333401
LBFGS: 37 04:43:30 8.750249 0.530715
LBFGS: 38 04:43:30 8.725928 0.685116
LBFGS: 39 04:43:30 8.702312 0.582260
LBFGS: 40 04:43:30 8.661515 0.399625
LBFGS: 41 04:43:30 8.643432 0.558474
LBFGS: 42 04:43:30 8.621201 0.367288
LBFGS: 43 04:43:30 8.614414 0.139424
LBFGS: 44 04:43:30 8.610785 0.137160
LBFGS: 45 04:43:30 8.608134 0.146375
LBFGS: 46 04:43:30 8.604928 0.119648
LBFGS: 47 04:43:30 8.599151 0.135424
LBFGS: 48 04:43:30 8.594063 0.147913
LBFGS: 49 04:43:30 8.589493 0.153840
LBFGS: 50 04:43:30 8.587274 0.088460
LBFGS: 51 04:43:30 8.584633 0.093750
LBFGS: 52 04:43:30 8.580239 0.140870
LBFGS: 53 04:43:30 8.572938 0.254272
LBFGS: 54 04:43:30 8.563343 0.291885
LBFGS: 55 04:43:30 8.554117 0.196557
LBFGS: 56 04:43:30 8.547597 0.129064
LBFGS: 57 04:43:30 8.542086 0.128020
LBFGS: 58 04:43:30 8.535432 0.098202
LBFGS: 59 04:43:30 8.533622 0.127672
LBFGS: 60 04:43:30 8.527487 0.116729
LBFGS: 61 04:43:30 8.523863 0.121762
LBFGS: 62 04:43:30 8.519229 0.130541
LBFGS: 63 04:43:30 8.515424 0.101902
LBFGS: 64 04:43:30 8.511240 0.212223
LBFGS: 65 04:43:30 8.507967 0.266593
LBFGS: 66 04:43:30 8.503903 0.237734
LBFGS: 67 04:43:30 8.497575 0.162253
LBFGS: 68 04:43:30 8.485434 0.202203
LBFGS: 69 04:43:30 8.466738 0.215895
LBFGS: 70 04:43:30 8.467607 0.334764
LBFGS: 71 04:43:30 8.454037 0.106310
LBFGS: 72 04:43:30 8.448980 0.119721
LBFGS: 73 04:43:30 8.446550 0.099221
LBFGS: 74 04:43:30 8.444705 0.056244
LBFGS: 75 04:43:30 8.443403 0.038831
LBFGS: 76 04:43:30 8.442646 0.054772
LBFGS: 77 04:43:30 8.442114 0.061370
LBFGS: 78 04:43:30 8.440960 0.058800
LBFGS: 79 04:43:30 8.439820 0.048198
LBFGS: 80 04:43:30 8.438600 0.051251
LBFGS: 81 04:43:30 8.437429 0.054130
LBFGS: 82 04:43:30 8.435695 0.067234
LBFGS: 83 04:43:30 8.431957 0.085678
LBFGS: 84 04:43:30 8.423485 0.133240
LBFGS: 85 04:43:30 8.413846 0.207812
LBFGS: 86 04:43:30 8.404849 0.178747
LBFGS: 87 04:43:30 8.385339 0.169017
LBFGS: 88 04:43:30 8.386849 0.187645
LBFGS: 89 04:43:30 8.371078 0.118124
LBFGS: 90 04:43:30 8.368801 0.094222
LBFGS: 91 04:43:30 8.366226 0.066960
LBFGS: 92 04:43:30 8.361680 0.054964
LBFGS: 93 04:43:30 8.360631 0.047342
LBFGS: 94 04:43:30 8.359692 0.024179
LBFGS: 95 04:43:30 8.359361 0.015549
LBFGS: 96 04:43:30 8.359163 0.014284
LBFGS: 97 04:43:30 8.359102 0.015615
LBFGS: 98 04:43:30 8.359048 0.015492
LBFGS: 99 04:43:30 8.358986 0.014214
LBFGS: 100 04:43:30 8.358921 0.013159
/opt/hostedtoolcache/Python/3.12.9/x64/lib/python3.12/site-packages/ase/io/extxyz.py:311: UserWarning: Skipping unhashable information adsorbate_info
warnings.warn('Skipping unhashable information '
Reading a trajectory#
identifier = "toy_c3h8_relax.extxyz"
# the `index` argument corresponds to what frame of the trajectory to read in, specifiying ":" reads in the full trajectory.
traj = ase.io.read(f"data/{identifier}", index=":")
Viewing a trajectory#
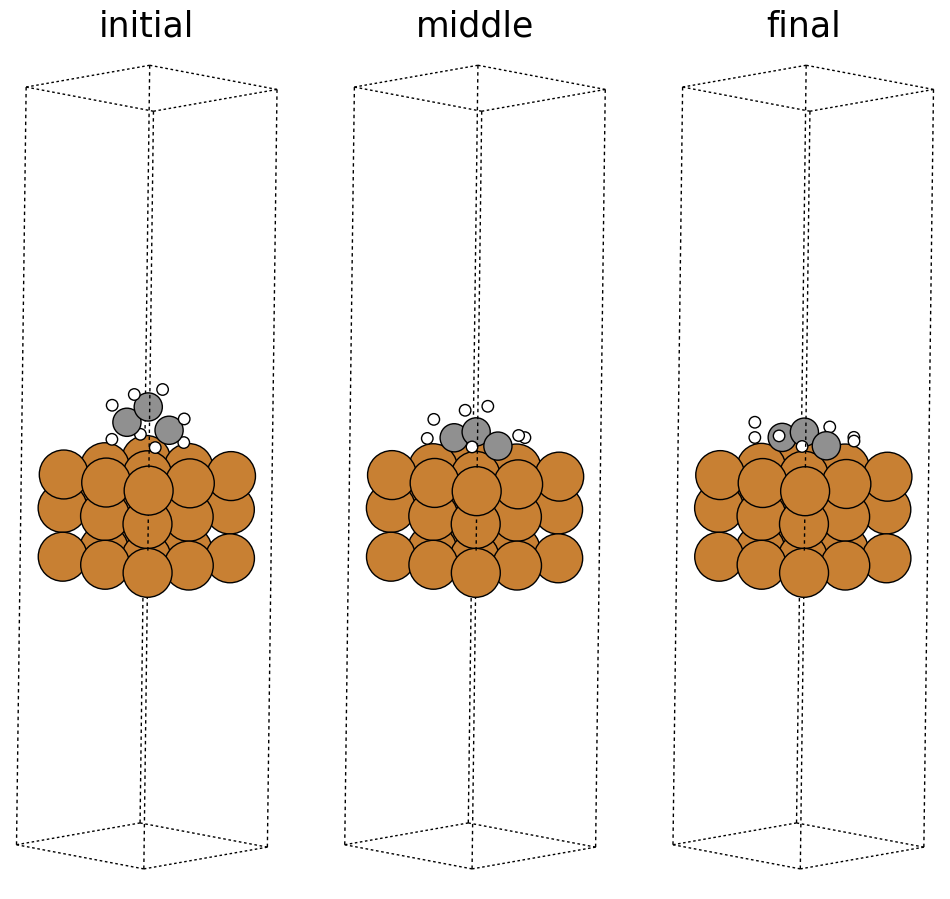
Below we visualize the initial, middle, and final steps in the structural relaxation trajectory from above. Copper atoms in the surface are colored orange, the propane adsorbate on the surface has grey colored carbon atoms and white colored hydrogen atoms. The adsorbate’s structure changes during the simulation and you can see how it relaxes on the surface. In this case, the relaxation looks normal; however, there can be instances where the adsorbate flies away (desorbs) from the surface or the adsorbate can break apart (dissociation), which are hard to detect without visualization. Additionally, visualizations can be used as a quick sanity check to ensure the initial system is set up correctly and there are no major issues with the simulation.
fig, ax = plt.subplots(1, 3)
labels = ['initial', 'middle', 'final']
for i in range(3):
ax[i].axis('off')
ax[i].set_title(labels[i])
ase.visualize.plot.plot_atoms(traj[0],
ax[0],
radii=0.8,
rotation=("-75x, 45y, 10z"))
ase.visualize.plot.plot_atoms(traj[50],
ax[1],
radii=0.8,
rotation=("-75x, 45y, 10z"))
ase.visualize.plot.plot_atoms(traj[-1],
ax[2],
radii=0.8,
rotation=("-75x, 45y, 10z"))
<Axes: title={'center': 'final'}>
Data contents #
Here we take a closer look at what information is contained within these trajectories.
i_structure = traj[0]
i_structure
Atoms(symbols='Cu27C3H8', pbc=True, cell=[7.65796644025031, 7.65796644025031, 33.266996999999996], tags=..., constraint=FixAtoms(indices=[0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17]), calculator=SinglePointCalculator(...))
Atomic numbers#
numbers = i_structure.get_atomic_numbers()
print(numbers)
[29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29 29
29 29 29 6 6 6 1 1 1 1 1 1 1 1]
Atomic symbols#
symbols = np.array(i_structure.get_chemical_symbols())
print(symbols)
['Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu'
'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'Cu' 'C' 'C'
'C' 'H' 'H' 'H' 'H' 'H' 'H' 'H' 'H']
Unit cell#
The unit cell is the volume containing our system of interest. Express as a 3x3 array representing the directional vectors that make up the volume. Illustrated as the dashed box in the above visuals.
cell = np.array(i_structure.cell)
print(cell)
[[ 7.65796644 0. 0. ]
[ 0. 7.65796644 0. ]
[ 0. 0. 33.266997 ]]
Periodic boundary conditions (PBC)#
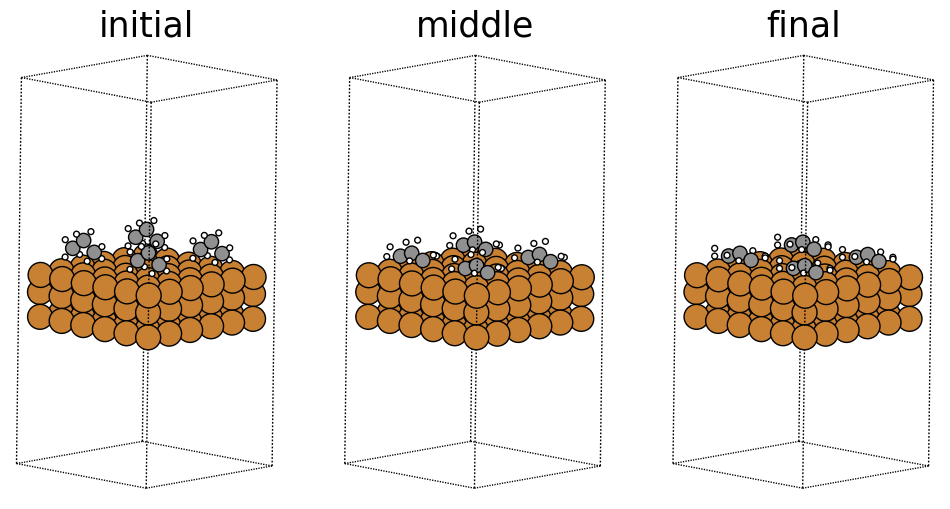
x,y,z boolean representing whether a unit cell repeats in the corresponding directions. The OC20 dataset sets this to [True, True, True], with a large enough vacuum layer above the surface such that a unit cell does not see itself in the z direction. Although the original structure shown above is what get’s passed into our models, the presence of PBC allows it to effectively repeat infinitely in the x and y directions. Below we visualize the same structure with a periodicity of 2 in all directions, what the model may effectively see.
pbc = i_structure.pbc
print(pbc)
[ True True True]
fig, ax = plt.subplots(1, 3)
labels = ['initial', 'middle', 'final']
for i in range(3):
ax[i].axis('off')
ax[i].set_title(labels[i])
ase.visualize.plot.plot_atoms(traj[0].repeat((2,2,1)),
ax[0],
radii=0.8,
rotation=("-75x, 45y, 10z"))
ase.visualize.plot.plot_atoms(traj[50].repeat((2,2,1)),
ax[1],
radii=0.8,
rotation=("-75x, 45y, 10z"))
ase.visualize.plot.plot_atoms(traj[-1].repeat((2,2,1)),
ax[2],
radii=0.8,
rotation=("-75x, 45y, 10z"))
<Axes: title={'center': 'final'}>
Fixed atoms constraint#
In reality, surfaces contain many, many more atoms beneath what we’ve illustrated as the surface. At an infinite depth, these subsurface atoms would look just like the bulk structure. We approximate a true surface by fixing the subsurface atoms into their “bulk” locations. This ensures that they cannot move at the “bottom” of the surface. If they could, this would throw off our calculations. Consistent with the above, we fix all atoms with tags=0, and denote them as “fixed”. All other atoms are considered “free”.
cons = i_structure.constraints[0]
print(cons, '\n')
# indices of fixed atoms
indices = cons.index
print(indices, '\n')
# fixed atoms correspond to tags = 0
print(tags[indices])
FixAtoms(indices=[0, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17])
[ 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17]
[0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0]
Adsorption energy#
The energy of the system is one of the properties of interest in the OC20 dataset. It’s important to note that absolute energies provide little value to researchers and must be referenced properly to be useful. The OC20 dataset references all it’s energies to the bare slab + gas references to arrive at adsorption energies. Adsorption energies are important in studying catalysts and their corresponding reaction rates. In addition to the structure relaxations of the OC20 dataset, bare slab and gas (N2, H2, H2O, CO) relaxations were carried out with DFT in order to calculate adsorption energies.
final_structure = traj[-1]
relaxed_energy = final_structure.get_potential_energy()
print(f'Relaxed absolute energy = {relaxed_energy} eV')
# Corresponding raw slab used in original adslab (adsorbate+slab) system.
raw_slab = fcc100("Cu", size=(3, 3, 3))
raw_slab.set_calculator(EMT())
raw_slab_energy = raw_slab.get_potential_energy()
print(f'Raw slab energy = {raw_slab_energy} eV')
adsorbate = Atoms("C3H8").get_chemical_symbols()
# For clarity, we define arbitrary gas reference energies here.
# A more detailed discussion of these calculations can be found in the corresponding paper's SI.
gas_reference_energies = {'H': .3, 'O': .45, 'C': .35, 'N': .50}
adsorbate_reference_energy = 0
for ads in adsorbate:
adsorbate_reference_energy += gas_reference_energies[ads]
print(f'Adsorbate reference energy = {adsorbate_reference_energy} eV\n')
adsorption_energy = relaxed_energy - raw_slab_energy - adsorbate_reference_energy
print(f'Adsorption energy: {adsorption_energy} eV')
Relaxed absolute energy = 8.358921451410891 eV
Raw slab energy = 8.127167122749576 eV
Adsorbate reference energy = 3.4499999999999993 eV
Adsorption energy: -3.2182456713386838 eV
/tmp/ipykernel_3934/2478225434.py:7: FutureWarning: Please use atoms.calc = calc
raw_slab.set_calculator(EMT())
Plot energy profile of toy trajectory#
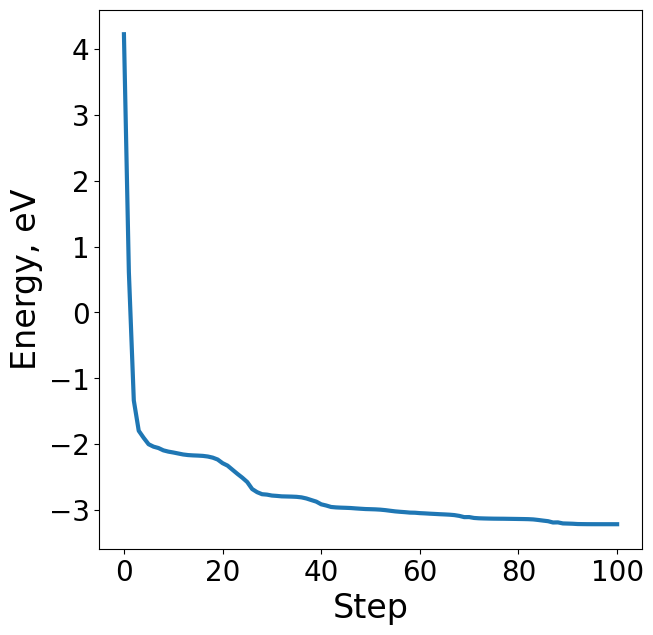
Plotting the energy profile of our trajectory is a good way to ensure nothing strange has occured. We expect to see a decreasing monotonic function. If the energy is consistently increasing or there’s multiple large spikes this could be a sign of some issues in the optimization. This is particularly useful for when analyzing ML-driven relaxations and whether they make general physical sense.
energies = [image.get_potential_energy() - raw_slab_energy - adsorbate_reference_energy for image in traj]
plt.figure(figsize=(7, 7))
plt.plot(range(len(energies)), energies, lw=3)
plt.xlabel("Step", fontsize=24)
plt.ylabel("Energy, eV", fontsize=24)
Text(0, 0.5, 'Energy, eV')
Force#
Forces are another important property of the OC20 dataset. Unlike datasets like QM9 which contain only ground state properties, the OC20 dataset contains per-atom forces necessary to carry out atomistic simulations. Physically, forces are the negative gradient of energy w.r.t atomic positions: \(F = -\frac{dE}{dx}\). Although not mandatory (depending on the application), maintaining this energy-force consistency is important for models that seek to make predictions on both properties.
The “apply_constraint” argument controls whether to apply system constraints to the forces. In the OC20 dataset, this controls whether to return forces for fixed atoms (apply_constraint=False) or return 0s (apply_constraint=True).
# Returning forces for all atoms - regardless of whether "fixed" or "free"
i_structure.get_forces(apply_constraint=False)
array([[-1.07900000e-05, -3.80000000e-06, 1.13560540e-01],
[ 0.00000000e+00, -4.29200000e-05, 1.13302410e-01],
[ 1.07900000e-05, -3.80000000e-06, 1.13560540e-01],
[-1.84600000e-05, 0.00000000e+00, 1.13543430e-01],
[ 0.00000000e+00, 0.00000000e+00, 1.13047800e-01],
[ 1.84600000e-05, -0.00000000e+00, 1.13543430e-01],
[-1.07900000e-05, 3.80000000e-06, 1.13560540e-01],
[-0.00000000e+00, 4.29200000e-05, 1.13302410e-01],
[ 1.07900000e-05, 3.80000000e-06, 1.13560540e-01],
[-1.10430500e-02, -2.53094000e-03, -4.84573700e-02],
[ 1.10430500e-02, -2.53094000e-03, -4.84573700e-02],
[-0.00000000e+00, -2.20890000e-04, -2.07827000e-03],
[-1.10430500e-02, 2.53094000e-03, -4.84573700e-02],
[ 1.10430500e-02, 2.53094000e-03, -4.84573700e-02],
[-0.00000000e+00, 2.20890000e-04, -2.07827000e-03],
[-3.49808000e-03, -0.00000000e+00, -7.85544000e-03],
[ 3.49808000e-03, -0.00000000e+00, -7.85544000e-03],
[-0.00000000e+00, -0.00000000e+00, -5.97640000e-04],
[-3.18144370e-01, -2.36420450e-01, -3.97089230e-01],
[ 0.00000000e+00, -2.18895316e+00, -2.74768262e+00],
[ 3.18144370e-01, -2.36420450e-01, -3.97089230e-01],
[-5.65980520e-01, 0.00000000e+00, -6.16046990e-01],
[ 0.00000000e+00, -0.00000000e+00, -4.47152822e+00],
[ 5.65980520e-01, 0.00000000e+00, -6.16046990e-01],
[-3.18144370e-01, 2.36420450e-01, -3.97089230e-01],
[-0.00000000e+00, 2.18895316e+00, -2.74768262e+00],
[ 3.18144370e-01, 2.36420450e-01, -3.97089230e-01],
[-0.00000000e+00, 0.00000000e+00, -3.96835355e+00],
[-0.00000000e+00, -3.64190926e+00, 5.71458646e+00],
[-0.00000000e+00, 3.64190926e+00, 5.71458646e+00],
[-2.18178516e+00, 0.00000000e+00, 1.67589182e+00],
[ 2.18178516e+00, 0.00000000e+00, 1.67589182e+00],
[ 0.00000000e+00, 2.46333681e+00, 1.78299828e+00],
[ 0.00000000e+00, -2.46333681e+00, 1.78299828e+00],
[ 6.18714050e+00, 2.26336330e-01, -5.99485570e-01],
[-6.18714050e+00, 2.26336330e-01, -5.99485570e-01],
[-6.18714050e+00, -2.26336330e-01, -5.99485570e-01],
[ 6.18714050e+00, -2.26336330e-01, -5.99485570e-01]])
# Applying the fixed atoms constraint to the forces
i_structure.get_forces(apply_constraint=True)
array([[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[ 0. , 0. , 0. ],
[-0.31814437, -0.23642045, -0.39708923],
[ 0. , -2.18895316, -2.74768262],
[ 0.31814437, -0.23642045, -0.39708923],
[-0.56598052, 0. , -0.61604699],
[ 0. , -0. , -4.47152822],
[ 0.56598052, 0. , -0.61604699],
[-0.31814437, 0.23642045, -0.39708923],
[-0. , 2.18895316, -2.74768262],
[ 0.31814437, 0.23642045, -0.39708923],
[-0. , 0. , -3.96835355],
[-0. , -3.64190926, 5.71458646],
[-0. , 3.64190926, 5.71458646],
[-2.18178516, 0. , 1.67589182],
[ 2.18178516, 0. , 1.67589182],
[ 0. , 2.46333681, 1.78299828],
[ 0. , -2.46333681, 1.78299828],
[ 6.1871405 , 0.22633633, -0.59948557],
[-6.1871405 , 0.22633633, -0.59948557],
[-6.1871405 , -0.22633633, -0.59948557],
[ 6.1871405 , -0.22633633, -0.59948557]])
Interacting with the OC20 datasets#
The OC20 datasets are stored in LMDBs. Here we show how to interact with the datasets directly in order to better understand the data. We use LmdbDataset to read in a directory of LMDB files or a single LMDB file.
from fairchem.core.datasets import LmdbDataset
# LmdbDataset is our custom Dataset method to read the lmdbs as Data objects. Note that we need to give the path to the folder containing lmdbs for S2EF
dataset = LmdbDataset({"src": "data/s2ef/train_100/"})
print("Size of the dataset created:", len(dataset))
print(dataset[0])
Size of the dataset created: 100
Data(edge_index=[2, 2964], y=6.282500615000004, pos=[86, 3], cell=[1, 3, 3], atomic_numbers=[86], natoms=86, cell_offsets=[2964, 3], force=[86, 3], fixed=[86], tags=[86], sid=[1], fid=[1], total_frames=74, id='0_0')
data = dataset[0]
data
Data(edge_index=[2, 2964], y=6.282500615000004, pos=[86, 3], cell=[1, 3, 3], atomic_numbers=[86], natoms=86, cell_offsets=[2964, 3], force=[86, 3], fixed=[86], tags=[86], sid=[1], fid=[1], total_frames=74, id='0_0')
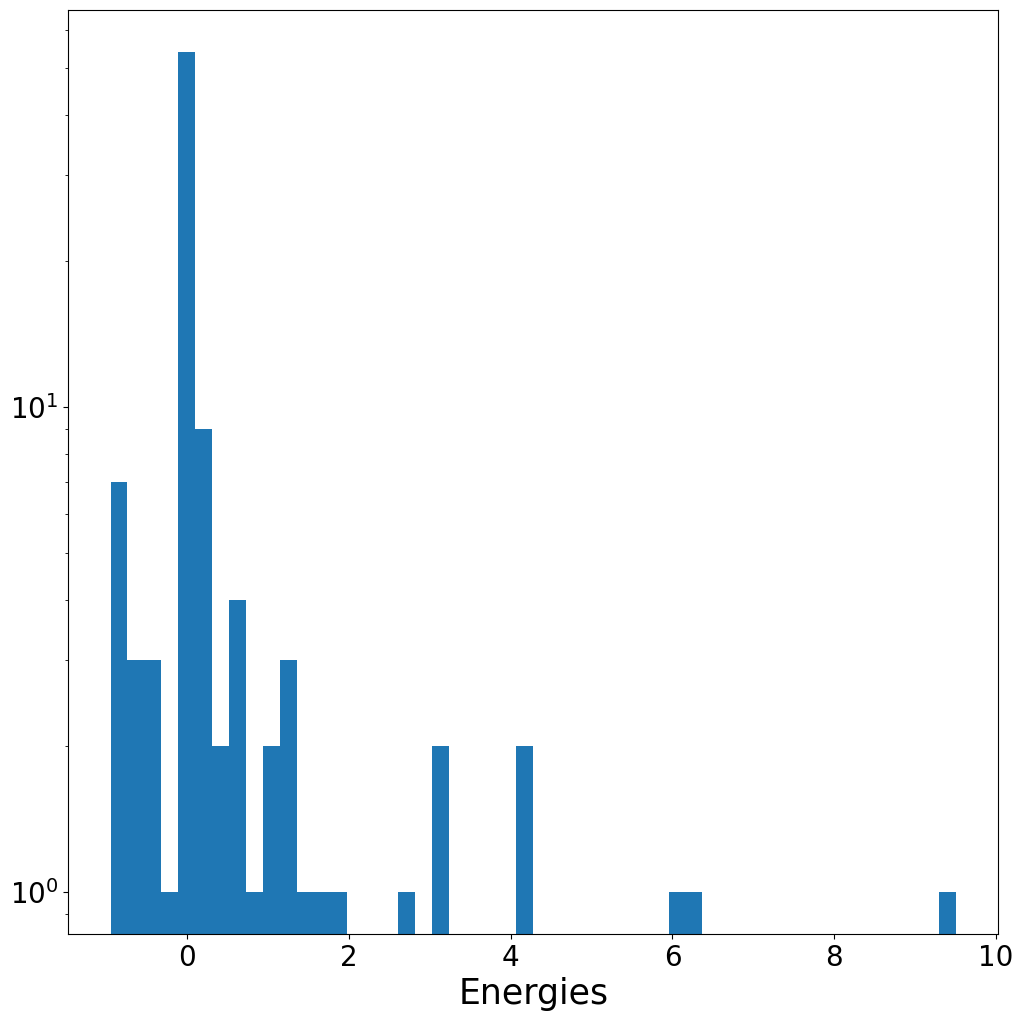
energies = torch.tensor([data.y for data in dataset])
energies
tensor([ 6.2825e+00, 4.1290e+00, 3.1451e+00, 3.0260e+00, 1.7921e+00,
1.6451e+00, 1.2257e+00, 1.2161e+00, 1.0712e+00, 7.4727e-01,
5.9575e-01, 5.7016e-01, 4.2819e-01, 3.1616e-01, 2.5283e-01,
2.2425e-01, 2.2346e-01, 2.0530e-01, 1.6090e-01, 1.1807e-01,
1.1691e-01, 9.1254e-02, 7.4997e-02, 6.3274e-02, 5.2782e-02,
4.8892e-02, 3.9609e-02, 3.1746e-02, 2.7179e-02, 2.7007e-02,
2.3709e-02, 1.8005e-02, 1.7676e-02, 1.4129e-02, 1.3162e-02,
1.1374e-02, 7.4124e-03, 7.7525e-03, 6.1224e-03, 5.2787e-03,
2.8587e-03, 1.1835e-04, -1.1200e-03, -1.3011e-03, -2.6812e-03,
-5.9202e-03, -6.1644e-03, -6.9261e-03, -9.1364e-03, -9.2114e-03,
-1.0665e-02, -1.3760e-02, -1.3588e-02, -1.4895e-02, -1.6190e-02,
-1.8660e-02, -1.4980e-02, -1.8880e-02, -2.0218e-02, -2.0559e-02,
-2.1013e-02, -2.2129e-02, -2.2748e-02, -2.3322e-02, -2.3382e-02,
-2.3865e-02, -2.3973e-02, -2.4196e-02, -2.4755e-02, -2.4951e-02,
-2.5078e-02, -2.5148e-02, -2.5257e-02, -2.5550e-02, 5.9721e+00,
9.5081e+00, 2.6373e+00, 4.0946e+00, 1.4385e+00, 1.2700e+00,
1.0081e+00, 5.3797e-01, 5.1462e-01, 2.8812e-01, 1.2429e-01,
-1.1352e-02, -2.2293e-01, -3.9102e-01, -4.3574e-01, -5.3142e-01,
-5.4777e-01, -6.3948e-01, -7.3816e-01, -8.2163e-01, -8.2526e-01,
-8.8313e-01, -8.8615e-01, -9.3446e-01, -9.5100e-01, -9.5168e-01])
plt.hist(energies, bins = 50)
plt.yscale("log")
plt.xlabel("Energies")
plt.show()
Additional Resources#
More helpful resources, tutorials, and documentation can be found at ASE’s webpage: https://wiki.fysik.dtu.dk/ase/index.html. We point to specific pages that may be of interest:
Interacting with Atoms Object: https://wiki.fysik.dtu.dk/ase/ase/atoms.html
Visualization: https://wiki.fysik.dtu.dk/ase/ase/visualize/visualize.html
Structure optimization: https://wiki.fysik.dtu.dk/ase/ase/optimize.html
More ASE Tutorials: https://wiki.fysik.dtu.dk/ase/tutorials/tutorials.html
Tasks#
In this section, we cover the different types of tasks the OC20 dataset presents and how to train and predict their corresponding models.
Structure to Energy and Forces (S2EF)
Initial Structure to Relaxed Energy (IS2RE)
Initial Structure to Relaxed Structure (IS2RS)
Tasks can be interrelated. The figure below illustrates several approaches to solving the IS2RE task:
(a) the traditional approach uses DFT along with an optimizer, such as BFGS or conjugate gradient, to iteratively update the atom positions until the relaxed structure and energy are found.
(b) using ML models trained to predict the energy and forces of a structure, S2EF can be used as a direct replacement for DFT.
(c) the relaxed structure could potentially be directly regressed from the initial structure and S2EF used to find the energy.
(d) directly compute the relaxed energy from the initial state.
NOTE The following sections are intended to demonstrate the inner workings of our codebase and what goes into running the various tasks. We do not recommend training to completion within a notebook setting. Please see the running on command line section for the preferred way to train/evaluate models.

Structure to Energy and Forces (S2EF) #
The S2EF task takes an atomic system as input and predicts the energy of the entire system and forces on each atom. This is our most general task, ultimately serving as a surrogate to DFT. A model that can perform well on this task can accelerate other applications like molecular dynamics and transitions tate calculations.
Steps for training an S2EF model#
Define or load a configuration (config), which includes the following
task
model
optimizer
dataset
trainer
Create a ForcesTrainer object
Train the model
Validate the model
For storage and compute reasons we use a very small subset of the OC20 S2EF dataset for this tutorial. Results will be considerably worse than presented in our paper.
Imports#
from fairchem.core.trainers import OCPTrainer
from fairchem.core.datasets import LmdbDataset
from fairchem.core import models
from fairchem.core.common import logger
from fairchem.core.common.utils import setup_logging, setup_imports
setup_logging()
setup_imports()
import numpy as np
import copy
import os
2025-03-29 04:43:38 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:359: (INFO): Project root: /home/runner/work/fairchem/fairchem/src/fairchem
/home/runner/work/fairchem/fairchem/src/fairchem/core/models/scn/spherical_harmonics.py:23: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
_Jd = torch.load(os.path.join(os.path.dirname(__file__), "Jd.pt"))
/home/runner/work/fairchem/fairchem/src/fairchem/core/models/equiformer_v2/wigner.py:10: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
_Jd = torch.load(os.path.join(os.path.dirname(__file__), "Jd.pt"))
/home/runner/work/fairchem/fairchem/src/fairchem/core/models/escn/so3.py:23: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
_Jd = torch.load(os.path.join(os.path.dirname(__file__), "Jd.pt"))
Dataset#
train_src = "data/s2ef/train_100"
val_src = "data/s2ef/val_20"
Normalize data#
If you wish to normalize the targets we must compute the mean and standard deviation for our energy values. Because forces are physically related by the negative gradient of energy, we use the same multiplicative energy factor for forces.
train_dataset = LmdbDataset({"src": train_src})
energies = []
for data in train_dataset:
energies.append(data.y)
mean = np.mean(energies)
stdev = np.std(energies)
Define the Config#
For this example, we will explicitly define the config. Default config yaml files can easily be loaded with the following build_config utility. Loading a yaml config is preferable when launching jobs from the command line. We have included a set of default configs for our best models’ here.
We will also use a scaling files found here. Lets download it locally,
%%bash
wget https://github.com/FAIR-Chem/fairchem/raw/main/configs/oc20/s2ef/all/gemnet/scaling_factors/gemnet-oc-large.pt
wget https://github.com/FAIR-Chem/fairchem/raw/main/configs/oc20/s2ef/all/gemnet/scaling_factors/gemnet-oc.pt
wget https://github.com/FAIR-Chem/fairchem/raw/main/configs/oc20/s2ef/all/gemnet/scaling_factors/gemnet-dT.json
--2025-03-29 04:43:38-- https://github.com/FAIR-Chem/fairchem/raw/main/configs/oc20/s2ef/all/gemnet
/scaling_factors/gemnet-oc-large.pt
Resolving github.com (github.com)...
140.82.116.3
Connecting to github.com (github.com)|140.82.116.3|:443...
connected.
HTTP request sent, awaiting response...
302 Found
Location: https://raw.githubusercontent.com/FAIR-Chem/fairchem/main/configs/oc20/s2ef/all/
gemnet/scaling_factors/gemnet-oc-large.pt [following]
--2025-03-29 04:43:38-- https://raw.githubusercontent.com/FAIR-Chem/fairchem/main/configs/oc20/s2ef
/all/gemnet/scaling_factors/gemnet-oc-large.pt
Resolving raw.githubusercontent.com (raw.githubuserco
ntent.com)...
185.199.109.133, 185.199.110.133, 185.199.111.133, ...
Connecting to raw.githubusercontent.com (raw.
githubusercontent.com)|185.199.109.133|:443...
connected.
HTTP request sent, awaiting response...
200 OK
Length:
27199 (27K) [application/octet-stream]
Saving to: ‘gemnet-oc-large.pt’
0K .......... .......... ...... 1
00% 92.2M=0s
2025-03-29 04:43:38 (92.2 MB/s) - ‘gemnet-oc-large.pt’ saved [27199/27199]
--2025-03-29 04:43:38-- https://github.com/FAIR-Chem/fairchem/raw/main/configs/oc20/s2ef/all/gemnet
/scaling_factors/gemnet-oc.pt
Resolving github.com (github.com)...
140.82.116.3
Connecting to github.com (github.com)|140.82.116.3|:443...
connected.
HTTP request sent, awaiting response...
302 Found
Location: https://raw.githubusercontent.com/FAIR-Chem/fairchem/main/configs/oc20/s2ef/all/
gemnet/scaling_factors/gemnet-oc.pt [following]
--2025-03-29 04:43:39-- https://raw.githubusercontent.com/FAIR-Chem/fairchem/main/configs/oc20/s2ef
/all/gemnet/scaling_factors/gemnet-oc.pt
Resolving raw.githubusercontent.com (raw.githubusercontent.
com)... 185.199.110.133, 185.199.111.133, 185.199.109.133, ...
Connecting to raw.githubusercontent.c
om (raw.githubusercontent.com)|185.199.110.133|:443... connected.
HTTP request sent, awaiting response...
200 OK
Length: 16963 (17K) [application/octet-stream]
Saving to: ‘gemnet-oc.pt’
0K .......... ...
... 100% 98.0M=0s
2025-03-29 04:43:39 (98.0 MB/s) - ‘gemnet-o
c.pt’ saved [16963/16963]
--2025-03-29 04:43:39-- https://github.com/FAIR-Chem/fairchem/raw/main/configs/oc20/s2ef/all/gemnet
/scaling_factors/gemnet-dT.json
Resolving github.com (github.com)...
140.82.116.3
Connecting to github.com (github.com)|140.82.116.3|:443...
connected.
HTTP request sent, awaiting response...
302 Found
Location: https://raw.githubusercontent.com/FAIR-Chem/fairchem/main/configs/oc20/s2ef/all/
gemnet/scaling_factors/gemnet-dT.json [following]
--2025-03-29 04:43:39-- https://raw.githubusercontent.com/FAIR-Chem/fairchem/main/configs/oc20/s2ef
/all/gemnet/scaling_factors/gemnet-dT.json
Resolving raw.githubusercontent.com (raw.githubuserconten
t.com)... 185.199.111.133, 185.199.108.133, 185.199.110.133, ...
Connecting to raw.githubusercontent
.com (raw.githubusercontent.com)|185.199.111.133|:443...
connected.
HTTP request sent, awaiting response...
200 OK
Length: 816 [text/plain]
Saving to: ‘gemnet-dT.json’
0K
100% 50.5M=0s
2025-03-29 04:43:39 (50.5 MB/s) - ‘gemnet-dT.json’ saved [816
/816]
Note - we only train for a single epoch with a reduced batch size (GPU memory constraints) for demonstration purposes, modify accordingly for full convergence.
# Task
task = {
'dataset': 'lmdb', # dataset used for the S2EF task
'description': 'Regressing to energies and forces for DFT trajectories from OCP',
'type': 'regression',
'metric': 'mae',
'labels': ['potential energy'],
'grad_input': 'atomic forces',
'train_on_free_atoms': True,
'eval_on_free_atoms': True
}
# Model
model = {
"name": "gemnet_oc",
"num_spherical": 7,
"num_radial": 128,
"num_blocks": 4,
"emb_size_atom": 64,
"emb_size_edge": 64,
"emb_size_trip_in": 64,
"emb_size_trip_out": 64,
"emb_size_quad_in": 32,
"emb_size_quad_out": 32,
"emb_size_aint_in": 64,
"emb_size_aint_out": 64,
"emb_size_rbf": 16,
"emb_size_cbf": 16,
"emb_size_sbf": 32,
"num_before_skip": 2,
"num_after_skip": 2,
"num_concat": 1,
"num_atom": 3,
"num_output_afteratom": 3,
"cutoff": 12.0,
"cutoff_qint": 12.0,
"cutoff_aeaint": 12.0,
"cutoff_aint": 12.0,
"max_neighbors": 30,
"max_neighbors_qint": 8,
"max_neighbors_aeaint": 20,
"max_neighbors_aint": 1000,
"rbf": {
"name": "gaussian"
},
"envelope": {
"name": "polynomial",
"exponent": 5
},
"cbf": {"name": "spherical_harmonics"},
"sbf": {"name": "legendre_outer"},
"extensive": True,
"output_init": "HeOrthogonal",
"activation": "silu",
"regress_forces": True,
"direct_forces": True,
"forces_coupled": False,
"quad_interaction": True,
"atom_edge_interaction": True,
"edge_atom_interaction": True,
"atom_interaction": True,
"num_atom_emb_layers": 2,
"num_global_out_layers": 2,
"qint_tags": [1, 2],
"scale_file": "./gemnet-oc.pt"
}
# Optimizer
optimizer = {
'batch_size': 1, # originally 32
'eval_batch_size': 1, # originally 32
'num_workers': 2,
'lr_initial': 5.e-4,
'optimizer': 'AdamW',
'optimizer_params': {"amsgrad": True},
'scheduler': "ReduceLROnPlateau",
'mode': "min",
'factor': 0.8,
'patience': 3,
'max_epochs': 1, # used for demonstration purposes
'force_coefficient': 100,
'ema_decay': 0.999,
'clip_grad_norm': 10,
'loss_energy': 'mae',
'loss_force': 'l2mae',
}
# Dataset
dataset = [
{'src': train_src,
'normalize_labels': True,
"target_mean": mean,
"target_std": stdev,
"grad_target_mean": 0.0,
"grad_target_std": stdev
}, # train set
{'src': val_src}, # val set (optional)
]
Create the trainer#
trainer = OCPTrainer(
task=task,
model=copy.deepcopy(model), # copied for later use, not necessary in practice.
dataset=dataset,
optimizer=optimizer,
outputs={},
loss_functions={},
evaluation_metrics={},
name="s2ef",
identifier="S2EF-example",
run_dir=".", # directory to save results if is_debug=False. Prediction files are saved here so be careful not to override!
is_debug=False, # if True, do not save checkpoint, logs, or results
print_every=5,
seed=0, # random seed to use
logger="tensorboard", # logger of choice (tensorboard and wandb supported)
local_rank=0,
amp=True, # use PyTorch Automatic Mixed Precision (faster training and less memory usage),
)
2025-03-29 04:43:39 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:1319: (WARNING): Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
2025-03-29 04:43:39 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:208: (INFO): amp: true
cmd:
checkpoint_dir: ./checkpoints/2025-03-29-04-43-44-S2EF-example
commit: core:b88b66e,experimental:NA
identifier: S2EF-example
logs_dir: ./logs/tensorboard/2025-03-29-04-43-44-S2EF-example
print_every: 5
results_dir: ./results/2025-03-29-04-43-44-S2EF-example
seed: 0
timestamp_id: 2025-03-29-04-43-44-S2EF-example
version: 0.1.dev1+gb88b66e
dataset:
format: lmdb
grad_target_mean: 0.0
grad_target_std: !!python/object/apply:numpy.core.multiarray.scalar
- &id001 !!python/object/apply:numpy.dtype
args:
- f8
- false
- true
state: !!python/tuple
- 3
- <
- null
- null
- null
- -1
- -1
- 0
- !!binary |
dPVlWhRA+D8=
key_mapping:
force: forces
y: energy
normalize_labels: true
src: data/s2ef/train_100
target_mean: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
zSXlDMrm3D8=
target_std: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
dPVlWhRA+D8=
transforms:
normalizer:
energy:
mean: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
zSXlDMrm3D8=
stdev: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
dPVlWhRA+D8=
forces:
mean: 0.0
stdev: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
dPVlWhRA+D8=
evaluation_metrics:
metrics:
energy:
- mae
forces:
- forcesx_mae
- forcesy_mae
- forcesz_mae
- mae
- cosine_similarity
- magnitude_error
misc:
- energy_forces_within_threshold
gp_gpus: null
gpus: 0
logger: tensorboard
loss_functions:
- energy:
coefficient: 1
fn: mae
- forces:
coefficient: 100
fn: l2mae
model:
activation: silu
atom_edge_interaction: true
atom_interaction: true
cbf:
name: spherical_harmonics
cutoff: 12.0
cutoff_aeaint: 12.0
cutoff_aint: 12.0
cutoff_qint: 12.0
direct_forces: true
edge_atom_interaction: true
emb_size_aint_in: 64
emb_size_aint_out: 64
emb_size_atom: 64
emb_size_cbf: 16
emb_size_edge: 64
emb_size_quad_in: 32
emb_size_quad_out: 32
emb_size_rbf: 16
emb_size_sbf: 32
emb_size_trip_in: 64
emb_size_trip_out: 64
envelope:
exponent: 5
name: polynomial
extensive: true
forces_coupled: false
max_neighbors: 30
max_neighbors_aeaint: 20
max_neighbors_aint: 1000
max_neighbors_qint: 8
name: gemnet_oc
num_after_skip: 2
num_atom: 3
num_atom_emb_layers: 2
num_before_skip: 2
num_blocks: 4
num_concat: 1
num_global_out_layers: 2
num_output_afteratom: 3
num_radial: 128
num_spherical: 7
output_init: HeOrthogonal
qint_tags:
- 1
- 2
quad_interaction: true
rbf:
name: gaussian
regress_forces: true
sbf:
name: legendre_outer
scale_file: ./gemnet-oc.pt
optim:
batch_size: 1
clip_grad_norm: 10
ema_decay: 0.999
eval_batch_size: 1
factor: 0.8
force_coefficient: 100
loss_energy: mae
loss_force: l2mae
lr_initial: 0.0005
max_epochs: 1
mode: min
num_workers: 2
optimizer: AdamW
optimizer_params:
amsgrad: true
patience: 3
scheduler: ReduceLROnPlateau
outputs:
energy:
level: system
forces:
eval_on_free_atoms: true
level: atom
train_on_free_atoms: true
relax_dataset: {}
slurm: {}
task:
dataset: lmdb
description: Regressing to energies and forces for DFT trajectories from OCP
eval_on_free_atoms: true
grad_input: atomic forces
labels:
- potential energy
metric: mae
train_on_free_atoms: true
type: regression
test_dataset: {}
trainer: s2ef
val_dataset:
src: data/s2ef/val_20
2025-03-29 04:43:39 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:557: (INFO): Loading model: gemnet_oc
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:568: (INFO): Loaded GemNetOC with 2596214 parameters.
/home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:37: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
scale_dict = torch.load(path)
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/logger.py:175: (WARNING): log_summary for Tensorboard not supported
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:358: (INFO): Loading dataset: lmdb
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/train_100')]'
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f2423bbbce0>, batch_size=1, drop_last=False
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/val_20')]'
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f2423be7ec0>, batch_size=1, drop_last=False
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy'] have been saved to: ./checkpoints/2025-03-29-04-43-44-S2EF-example/normalizers.pt
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy', 'forces'] have been saved to: ./checkpoints/2025-03-29-04-43-44-S2EF-example/normalizers.pt
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output energy: mean=0.45158625849998374, rmsd=1.5156444102461508.
2025-03-29 04:43:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output forces: mean=0.0, rmsd=1.5156444102461508.
Train the model#
trainer.train()
2025-03-29 04:43:46 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 3.09e+01, forcesx_mae: 3.30e-01, forcesy_mae: 3.57e-01, forcesz_mae: 4.72e-01, forces_mae: 3.87e-01, forces_cosine_similarity: 1.86e-02, forces_magnitude_error: 6.38e-01, loss: 7.25e+01, lr: 5.00e-04, epoch: 5.00e-02, step: 5.00e+00
2025-03-29 04:43:51 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 8.43e+00, forcesx_mae: 1.68e-01, forcesy_mae: 1.81e-01, forcesz_mae: 2.19e-01, forces_mae: 1.89e-01, forces_cosine_similarity: 4.28e-02, forces_magnitude_error: 2.69e-01, loss: 3.04e+01, lr: 5.00e-04, epoch: 1.00e-01, step: 1.00e+01
2025-03-29 04:43:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 4.58e+00, forcesx_mae: 1.44e-01, forcesy_mae: 2.21e-01, forcesz_mae: 2.22e-01, forces_mae: 1.95e-01, forces_cosine_similarity: 8.30e-02, forces_magnitude_error: 2.61e-01, loss: 2.73e+01, lr: 5.00e-04, epoch: 1.50e-01, step: 1.50e+01
2025-03-29 04:44:01 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 2.41e+01, forcesx_mae: 3.02e-01, forcesy_mae: 1.15e+00, forcesz_mae: 5.77e-01, forces_mae: 6.76e-01, forces_cosine_similarity: -1.29e-01, forces_magnitude_error: 1.09e+00, loss: 1.00e+02, lr: 5.00e-04, epoch: 2.00e-01, step: 2.00e+01
2025-03-29 04:44:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 9.50e+00, forcesx_mae: 2.23e-01, forcesy_mae: 4.82e-01, forcesz_mae: 3.30e-01, forces_mae: 3.45e-01, forces_cosine_similarity: 1.79e-01, forces_magnitude_error: 4.21e-01, loss: 4.95e+01, lr: 5.00e-04, epoch: 2.50e-01, step: 2.50e+01
2025-03-29 04:44:11 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 3.92e+00, forcesx_mae: 9.23e-02, forcesy_mae: 1.45e-01, forcesz_mae: 1.41e-01, forces_mae: 1.26e-01, forces_cosine_similarity: 1.35e-01, forces_magnitude_error: 1.37e-01, loss: 1.81e+01, lr: 5.00e-04, epoch: 3.00e-01, step: 3.00e+01
2025-03-29 04:44:16 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.27e+00, forcesx_mae: 9.02e-02, forcesy_mae: 1.01e-01, forcesz_mae: 1.37e-01, forces_mae: 1.09e-01, forces_cosine_similarity: 1.75e-01, forces_magnitude_error: 1.57e-01, loss: 1.53e+01, lr: 5.00e-04, epoch: 3.50e-01, step: 3.50e+01
2025-03-29 04:44:21 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 3.54e+00, forcesx_mae: 1.15e-01, forcesy_mae: 2.91e-01, forcesz_mae: 1.59e-01, forces_mae: 1.88e-01, forces_cosine_similarity: 1.06e-01, forces_magnitude_error: 2.78e-01, loss: 2.80e+01, lr: 5.00e-04, epoch: 4.00e-01, step: 4.00e+01
2025-03-29 04:44:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 5.46e+00, forcesx_mae: 2.24e-01, forcesy_mae: 4.34e-01, forcesz_mae: 2.30e-01, forces_mae: 2.96e-01, forces_cosine_similarity: 6.79e-02, forces_magnitude_error: 5.51e-01, loss: 3.59e+01, lr: 5.00e-04, epoch: 4.50e-01, step: 4.50e+01
2025-03-29 04:44:31 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.35e+00, forcesx_mae: 9.93e-02, forcesy_mae: 1.12e-01, forcesz_mae: 1.72e-01, forces_mae: 1.28e-01, forces_cosine_similarity: 2.90e-01, forces_magnitude_error: 2.32e-01, loss: 1.78e+01, lr: 5.00e-04, epoch: 5.00e-01, step: 5.00e+01
2025-03-29 04:44:37 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 3.67e+00, forcesx_mae: 1.29e-01, forcesy_mae: 2.21e-01, forcesz_mae: 1.52e-01, forces_mae: 1.67e-01, forces_cosine_similarity: 8.87e-02, forces_magnitude_error: 1.98e-01, loss: 2.35e+01, lr: 5.00e-04, epoch: 5.50e-01, step: 5.50e+01
2025-03-29 04:44:42 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.80e+00, forcesx_mae: 1.35e-01, forcesy_mae: 1.67e-01, forcesz_mae: 1.72e-01, forces_mae: 1.58e-01, forces_cosine_similarity: 1.41e-01, forces_magnitude_error: 2.35e-01, loss: 1.84e+01, lr: 5.00e-04, epoch: 6.00e-01, step: 6.00e+01
2025-03-29 04:44:47 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 2.54e+00, forcesx_mae: 1.12e-01, forcesy_mae: 1.63e-01, forcesz_mae: 1.58e-01, forces_mae: 1.44e-01, forces_cosine_similarity: 2.15e-01, forces_magnitude_error: 1.86e-01, loss: 1.87e+01, lr: 5.00e-04, epoch: 6.50e-01, step: 6.50e+01
2025-03-29 04:44:52 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 5.65e+00, forcesx_mae: 8.02e-02, forcesy_mae: 7.68e-02, forcesz_mae: 1.02e-01, forces_mae: 8.65e-02, forces_cosine_similarity: 2.34e-01, forces_magnitude_error: 1.13e-01, loss: 1.31e+01, lr: 5.00e-04, epoch: 7.00e-01, step: 7.00e+01
2025-03-29 04:44:57 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 3.50e+00, forcesx_mae: 1.66e-01, forcesy_mae: 1.86e-01, forcesz_mae: 2.47e-01, forces_mae: 2.00e-01, forces_cosine_similarity: 1.88e-01, forces_magnitude_error: 3.13e-01, loss: 2.87e+01, lr: 5.00e-04, epoch: 7.50e-01, step: 7.50e+01
2025-03-29 04:45:02 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 3.48e+00, forcesx_mae: 9.29e-02, forcesy_mae: 1.61e-01, forcesz_mae: 1.26e-01, forces_mae: 1.27e-01, forces_cosine_similarity: 1.74e-01, forces_magnitude_error: 1.60e-01, loss: 1.78e+01, lr: 5.00e-04, epoch: 8.00e-01, step: 8.00e+01
2025-03-29 04:45:07 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 4.68e+00, forcesx_mae: 1.63e-01, forcesy_mae: 2.52e-01, forcesz_mae: 2.53e-01, forces_mae: 2.23e-01, forces_cosine_similarity: 1.71e-01, forces_magnitude_error: 3.72e-01, loss: 3.41e+01, lr: 5.00e-04, epoch: 8.50e-01, step: 8.50e+01
2025-03-29 04:45:12 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.49e+00, forcesx_mae: 4.96e-02, forcesy_mae: 7.33e-02, forcesz_mae: 5.31e-02, forces_mae: 5.86e-02, forces_cosine_similarity: 1.68e-01, forces_magnitude_error: 7.36e-02, loss: 7.56e+00, lr: 5.00e-04, epoch: 9.00e-01, step: 9.00e+01
2025-03-29 04:45:18 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 2.72e+00, forcesx_mae: 2.11e-01, forcesy_mae: 2.87e-01, forcesz_mae: 3.33e-01, forces_mae: 2.77e-01, forces_cosine_similarity: 2.29e-01, forces_magnitude_error: 5.15e-01, loss: 3.04e+01, lr: 5.00e-04, epoch: 9.50e-01, step: 9.50e+01
2025-03-29 04:45:22 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 7.20e-01, forcesx_mae: 3.24e-02, forcesy_mae: 3.54e-02, forcesz_mae: 5.30e-02, forces_mae: 4.03e-02, forces_cosine_similarity: 2.61e-01, forces_magnitude_error: 5.69e-02, loss: 5.80e+00, lr: 5.00e-04, epoch: 1.00e+00, step: 1.00e+02
2025-03-29 04:45:23 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/20 [00:00<?, ?it/s]
device 0: 5%|▌ | 1/20 [00:00<00:07, 2.52it/s]
device 0: 10%|█ | 2/20 [00:00<00:05, 3.33it/s]
device 0: 15%|█▌ | 3/20 [00:00<00:04, 3.82it/s]
device 0: 20%|██ | 4/20 [00:01<00:03, 4.22it/s]
device 0: 25%|██▌ | 5/20 [00:01<00:03, 4.48it/s]
device 0: 30%|███ | 6/20 [00:01<00:03, 4.61it/s]
device 0: 35%|███▌ | 7/20 [00:01<00:02, 4.66it/s]
device 0: 40%|████ | 8/20 [00:01<00:02, 4.77it/s]
device 0: 45%|████▌ | 9/20 [00:02<00:02, 4.86it/s]
device 0: 50%|█████ | 10/20 [00:02<00:02, 4.92it/s]
device 0: 55%|█████▌ | 11/20 [00:02<00:01, 4.96it/s]
device 0: 60%|██████ | 12/20 [00:02<00:01, 4.92it/s]
device 0: 65%|██████▌ | 13/20 [00:02<00:01, 4.99it/s]
device 0: 70%|███████ | 14/20 [00:03<00:01, 5.04it/s]
device 0: 75%|███████▌ | 15/20 [00:03<00:00, 5.06it/s]
device 0: 80%|████████ | 16/20 [00:03<00:00, 5.10it/s]
device 0: 85%|████████▌ | 17/20 [00:03<00:00, 4.99it/s]
device 0: 90%|█████████ | 18/20 [00:03<00:00, 5.07it/s]
device 0: 95%|█████████▌| 19/20 [00:04<00:00, 5.11it/s]
device 0: 100%|██████████| 20/20 [00:04<00:00, 5.13it/s]
device 0: 100%|██████████| 20/20 [00:04<00:00, 4.71it/s]
2025-03-29 04:45:27 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 9.1440, forcesx_mae: 0.3023, forcesy_mae: 0.2598, forcesz_mae: 0.4722, forces_mae: 0.3448, forces_cosine_similarity: 0.0188, forces_magnitude_error: 0.4904, loss: 53.1097, epoch: 1.0000
Validate the model#
Load the best checkpoint#
The checkpoints directory contains two checkpoint files:
best_checkpoint.pt- Model parameters corresponding to the best val performance during training. Used for predictions.checkpoint.pt- Model parameters and optimizer settings for the latest checkpoint. Used to continue training.
# The `best_checpoint.pt` file contains the checkpoint with the best val performance
checkpoint_path = os.path.join(trainer.config["cmd"]["checkpoint_dir"], "best_checkpoint.pt")
checkpoint_path
'./checkpoints/2025-03-29-04-43-44-S2EF-example/best_checkpoint.pt'
# Append the dataset with the test set. We use the same val set for demonstration.
# Dataset
dataset.append(
{'src': val_src}, # test set (optional)
)
dataset
[{'src': 'data/s2ef/train_100',
'normalize_labels': True,
'target_mean': 0.45158625849998374,
'target_std': 1.5156444102461508,
'grad_target_mean': 0.0,
'grad_target_std': 1.5156444102461508},
{'src': 'data/s2ef/val_20'},
{'src': 'data/s2ef/val_20'}]
pretrained_trainer = OCPTrainer(
task=task,
model=copy.deepcopy(model), # copied for later use, not necessary in practice.
dataset=dataset,
optimizer=optimizer,
outputs={},
loss_functions={},
evaluation_metrics={},
name="s2ef",
identifier="S2EF-val-example",
run_dir="./", # directory to save results if is_debug=False. Prediction files are saved here so be careful not to override!
is_debug=False, # if True, do not save checkpoint, logs, or results
print_every=5,
seed=0, # random seed to use
logger="tensorboard", # logger of choice (tensorboard and wandb supported)
local_rank=0,
amp=True, # use PyTorch Automatic Mixed Precision (faster training and less memory usage),
)
pretrained_trainer.load_checkpoint(checkpoint_path=checkpoint_path)
2025-03-29 04:45:27 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:1319: (WARNING): Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
2025-03-29 04:45:27 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:208: (INFO): amp: true
cmd:
checkpoint_dir: ./checkpoints/2025-03-29-04-45-52-S2EF-val-example
commit: core:b88b66e,experimental:NA
identifier: S2EF-val-example
logs_dir: ./logs/tensorboard/2025-03-29-04-45-52-S2EF-val-example
print_every: 5
results_dir: ./results/2025-03-29-04-45-52-S2EF-val-example
seed: 0
timestamp_id: 2025-03-29-04-45-52-S2EF-val-example
version: 0.1.dev1+gb88b66e
dataset:
format: lmdb
grad_target_mean: 0.0
grad_target_std: !!python/object/apply:numpy.core.multiarray.scalar
- &id001 !!python/object/apply:numpy.dtype
args:
- f8
- false
- true
state: !!python/tuple
- 3
- <
- null
- null
- null
- -1
- -1
- 0
- !!binary |
dPVlWhRA+D8=
key_mapping:
force: forces
y: energy
normalize_labels: true
src: data/s2ef/train_100
target_mean: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
zSXlDMrm3D8=
target_std: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
dPVlWhRA+D8=
transforms:
normalizer:
energy:
mean: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
zSXlDMrm3D8=
stdev: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
dPVlWhRA+D8=
forces:
mean: 0.0
stdev: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
dPVlWhRA+D8=
evaluation_metrics:
metrics:
energy:
- mae
forces:
- forcesx_mae
- forcesy_mae
- forcesz_mae
- mae
- cosine_similarity
- magnitude_error
misc:
- energy_forces_within_threshold
gp_gpus: null
gpus: 0
logger: tensorboard
loss_functions:
- energy:
coefficient: 1
fn: mae
- forces:
coefficient: 100
fn: l2mae
model:
activation: silu
atom_edge_interaction: true
atom_interaction: true
cbf:
name: spherical_harmonics
cutoff: 12.0
cutoff_aeaint: 12.0
cutoff_aint: 12.0
cutoff_qint: 12.0
direct_forces: true
edge_atom_interaction: true
emb_size_aint_in: 64
emb_size_aint_out: 64
emb_size_atom: 64
emb_size_cbf: 16
emb_size_edge: 64
emb_size_quad_in: 32
emb_size_quad_out: 32
emb_size_rbf: 16
emb_size_sbf: 32
emb_size_trip_in: 64
emb_size_trip_out: 64
envelope:
exponent: 5
name: polynomial
extensive: true
forces_coupled: false
max_neighbors: 30
max_neighbors_aeaint: 20
max_neighbors_aint: 1000
max_neighbors_qint: 8
name: gemnet_oc
num_after_skip: 2
num_atom: 3
num_atom_emb_layers: 2
num_before_skip: 2
num_blocks: 4
num_concat: 1
num_global_out_layers: 2
num_output_afteratom: 3
num_radial: 128
num_spherical: 7
output_init: HeOrthogonal
qint_tags:
- 1
- 2
quad_interaction: true
rbf:
name: gaussian
regress_forces: true
sbf:
name: legendre_outer
scale_file: ./gemnet-oc.pt
optim:
batch_size: 1
clip_grad_norm: 10
ema_decay: 0.999
eval_batch_size: 1
factor: 0.8
force_coefficient: 100
loss_energy: mae
loss_force: l2mae
lr_initial: 0.0005
max_epochs: 1
mode: min
num_workers: 2
optimizer: AdamW
optimizer_params:
amsgrad: true
patience: 3
scheduler: ReduceLROnPlateau
outputs:
energy:
level: system
forces:
eval_on_free_atoms: true
level: atom
train_on_free_atoms: true
relax_dataset: {}
slurm: {}
task:
dataset: lmdb
description: Regressing to energies and forces for DFT trajectories from OCP
eval_on_free_atoms: true
grad_input: atomic forces
labels:
- potential energy
metric: mae
train_on_free_atoms: true
type: regression
test_dataset:
src: data/s2ef/val_20
trainer: s2ef
val_dataset:
src: data/s2ef/val_20
2025-03-29 04:45:27 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:557: (INFO): Loading model: gemnet_oc
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:568: (INFO): Loaded GemNetOC with 2596214 parameters.
/home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:37: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
scale_dict = torch.load(path)
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/logger.py:175: (WARNING): log_summary for Tensorboard not supported
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:358: (INFO): Loading dataset: lmdb
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/train_100')]'
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d8a7a40>, batch_size=1, drop_last=False
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/val_20')]'
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f2423b65ca0>, batch_size=1, drop_last=False
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/val_20')]'
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d735700>, batch_size=1, drop_last=False
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy'] have been saved to: ./checkpoints/2025-03-29-04-45-52-S2EF-val-example/normalizers.pt
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy', 'forces'] have been saved to: ./checkpoints/2025-03-29-04-45-52-S2EF-val-example/normalizers.pt
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output energy: mean=0.45158625849998374, rmsd=1.5156444102461508.
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output forces: mean=0.0, rmsd=1.5156444102461508.
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:606: (INFO): Loading checkpoint from: ./checkpoints/2025-03-29-04-43-44-S2EF-example/best_checkpoint.pt
/home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:607: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
checkpoint = torch.load(checkpoint_path, map_location=map_location)
Run on the test set#
# make predictions on the existing test_loader
predictions = pretrained_trainer.predict(pretrained_trainer.test_loader, results_file="s2ef_results", disable_tqdm=False)
2025-03-29 04:45:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:445: (INFO): Predicting on test.
device 0: 0%| | 0/20 [00:00<?, ?it/s]
device 0: 5%|▌ | 1/20 [00:00<00:07, 2.59it/s]
device 0: 10%|█ | 2/20 [00:00<00:05, 3.27it/s]
device 0: 15%|█▌ | 3/20 [00:00<00:04, 3.84it/s]
device 0: 20%|██ | 4/20 [00:01<00:03, 4.29it/s]
device 0: 25%|██▌ | 5/20 [00:01<00:03, 4.58it/s]
device 0: 30%|███ | 6/20 [00:01<00:02, 4.69it/s]
device 0: 35%|███▌ | 7/20 [00:01<00:02, 4.73it/s]
device 0: 40%|████ | 8/20 [00:01<00:02, 4.85it/s]
device 0: 45%|████▌ | 9/20 [00:02<00:02, 4.89it/s]
device 0: 50%|█████ | 10/20 [00:02<00:02, 4.96it/s]
device 0: 55%|█████▌ | 11/20 [00:02<00:01, 5.05it/s]
device 0: 60%|██████ | 12/20 [00:02<00:01, 5.03it/s]
device 0: 65%|██████▌ | 13/20 [00:02<00:01, 5.10it/s]
device 0: 70%|███████ | 14/20 [00:02<00:01, 5.14it/s]
device 0: 75%|███████▌ | 15/20 [00:03<00:00, 5.14it/s]
device 0: 80%|████████ | 16/20 [00:03<00:00, 5.19it/s]
device 0: 85%|████████▌ | 17/20 [00:03<00:00, 5.22it/s]
device 0: 90%|█████████ | 18/20 [00:03<00:00, 5.15it/s]
device 0: 95%|█████████▌| 19/20 [00:03<00:00, 5.20it/s]
device 0: 100%|██████████| 20/20 [00:04<00:00, 5.20it/s]
device 0: 100%|██████████| 20/20 [00:04<00:00, 4.78it/s]
2025-03-29 04:45:32 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:1006: (INFO): Writing results to ./results/2025-03-29-04-45-52-S2EF-val-example/s2ef_s2ef_results.npz
energies = predictions["energy"]
forces = predictions["forces"]
Initial Structure to Relaxed Energy (IS2RE) #
The IS2RE task predicts the relaxed energy (energy of the relaxed state) given the initial state of a system. One approach to this is by training a regression model mapping the initial structure to the relaxed energy. We call this the direct approach to the IS2RE task.
An alternative is to perform a structure relaxation using an S2EF model to obtain the relaxed state and compute the energy of that state (see the IS2RS task below for details about relaxation).
Steps for training an IS2RE model#
Define or load a configuration (config), which includes the following
task
model
optimizer
dataset
trainer
Create an EnergyTrainer object
Train the model
Validate the model
Imports#
from fairchem.core.trainers import OCPTrainer
from fairchem.core.datasets import LmdbDataset
from fairchem.core import models
from fairchem.core.common import logger
from fairchem.core.common.utils import setup_logging
setup_logging()
import numpy as np
import copy
import os
Dataset#
train_src = "data/is2re/train_100/data.lmdb"
val_src = "data/is2re/val_20/data.lmdb"
Normalize data#
If you wish to normalize the targets we must compute the mean and standard deviation for our energy values.
train_dataset = LmdbDataset({"src": train_src})
energies = []
for data in train_dataset:
energies.append(data.y_relaxed)
mean = np.mean(energies)
stdev = np.std(energies)
Define the Config#
For this example, we will explicitly define the config; however, a set of default configs can be found here. Default config yaml files can easily be loaded with the following utility. Loading a yaml config is preferrable when launching jobs from the command line. We have included our best models’ config files here for reference.
Note - we only train for a single epoch with a reduced batch size (GPU memory constraints) for demonstration purposes, modify accordingly for full convergence.
# Task
task = {
"dataset": "single_point_lmdb",
"description": "Relaxed state energy prediction from initial structure.",
"type": "regression",
"metric": "mae",
"labels": ["relaxed energy"],
}
# Model
model = {
'name': 'gemnet_t',
"num_spherical": 7,
"num_radial": 64,
"num_blocks": 5,
"emb_size_atom": 256,
"emb_size_edge": 512,
"emb_size_trip": 64,
"emb_size_rbf": 16,
"emb_size_cbf": 16,
"emb_size_bil_trip": 64,
"num_before_skip": 1,
"num_after_skip": 2,
"num_concat": 1,
"num_atom": 3,
"cutoff": 6.0,
"max_neighbors": 50,
"rbf": {"name": "gaussian"},
"envelope": {
"name": "polynomial",
"exponent": 5,
},
"cbf": {"name": "spherical_harmonics"},
"extensive": True,
"otf_graph": False,
"output_init": "HeOrthogonal",
"activation": "silu",
"regress_forces": False,
"direct_forces": False,
}
# Optimizer
optimizer = {
'batch_size': 1, # originally 32
'eval_batch_size': 1, # originally 32
'num_workers': 2,
'lr_initial': 1.e-4,
'optimizer': 'AdamW',
'optimizer_params': {"amsgrad": True},
'scheduler': "ReduceLROnPlateau",
'mode': "min",
'factor': 0.8,
'patience': 3,
'max_epochs': 1, # used for demonstration purposes
'ema_decay': 0.999,
'clip_grad_norm': 10,
'loss_energy': 'mae',
}
# Dataset
dataset = [
{'src': train_src,
'normalize_labels': True,
'target_mean': mean,
'target_std': stdev,
}, # train set
{'src': val_src}, # val set (optional)
]
###Create EnergyTrainer
energy_trainer = OCPTrainer(
task=task,
model=copy.deepcopy(model), # copied for later use, not necessary in practice.
dataset=dataset,
optimizer=optimizer,
outputs={},
loss_functions={},
evaluation_metrics={},
name="is2re",
identifier="IS2RE-example",
run_dir="./", # directory to save results if is_debug=False. Prediction files are saved here so be careful not to override!
is_debug=False, # if True, do not save checkpoint, logs, or results
print_every=5,
seed=0, # random seed to use
logger="tensorboard", # logger of choice (tensorboard and wandb supported)
local_rank=0,
amp=True, # use PyTorch Automatic Mixed Precision (faster training and less memory usage),
)
2025-03-29 04:45:32 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:1319: (WARNING): Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
2025-03-29 04:45:32 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:208: (INFO): amp: true
cmd:
checkpoint_dir: ./checkpoints/2025-03-29-04-45-52-IS2RE-example
commit: core:b88b66e,experimental:NA
identifier: IS2RE-example
logs_dir: ./logs/tensorboard/2025-03-29-04-45-52-IS2RE-example
print_every: 5
results_dir: ./results/2025-03-29-04-45-52-IS2RE-example
seed: 0
timestamp_id: 2025-03-29-04-45-52-IS2RE-example
version: 0.1.dev1+gb88b66e
dataset:
format: single_point_lmdb
key_mapping:
y_relaxed: energy
normalize_labels: true
src: data/is2re/train_100/data.lmdb
target_mean: !!python/object/apply:numpy.core.multiarray.scalar
- &id001 !!python/object/apply:numpy.dtype
args:
- f8
- false
- true
state: !!python/tuple
- 3
- <
- null
- null
- null
- -1
- -1
- 0
- !!binary |
MjyJzgpQ978=
target_std: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
PnyyzMtk/T8=
transforms:
normalizer:
energy:
mean: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
MjyJzgpQ978=
stdev: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
PnyyzMtk/T8=
forces:
mean: 0
stdev: 1
evaluation_metrics:
metrics:
energy:
- mae
- mse
- energy_within_threshold
gp_gpus: null
gpus: 0
logger: tensorboard
loss_functions:
- energy:
coefficient: 1
fn: mae
model:
activation: silu
cbf:
name: spherical_harmonics
cutoff: 6.0
direct_forces: false
emb_size_atom: 256
emb_size_bil_trip: 64
emb_size_cbf: 16
emb_size_edge: 512
emb_size_rbf: 16
emb_size_trip: 64
envelope:
exponent: 5
name: polynomial
extensive: true
max_neighbors: 50
name: gemnet_t
num_after_skip: 2
num_atom: 3
num_before_skip: 1
num_blocks: 5
num_concat: 1
num_radial: 64
num_spherical: 7
otf_graph: false
output_init: HeOrthogonal
rbf:
name: gaussian
regress_forces: false
optim:
batch_size: 1
clip_grad_norm: 10
ema_decay: 0.999
eval_batch_size: 1
factor: 0.8
loss_energy: mae
lr_initial: 0.0001
max_epochs: 1
mode: min
num_workers: 2
optimizer: AdamW
optimizer_params:
amsgrad: true
patience: 3
scheduler: ReduceLROnPlateau
outputs:
energy:
level: system
relax_dataset: {}
slurm: {}
task:
dataset: single_point_lmdb
description: Relaxed state energy prediction from initial structure.
labels:
- relaxed energy
metric: mae
type: regression
test_dataset: {}
trainer: is2re
val_dataset:
src: data/is2re/val_20/data.lmdb
2025-03-29 04:45:32 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:557: (INFO): Loading model: gemnet_t
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:568: (INFO): Loaded GemNetT with 22774037 parameters.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/logger.py:175: (WARNING): log_summary for Tensorboard not supported
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:358: (INFO): Loading dataset: single_point_lmdb
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/is2re/train_100/data.lmdb')]'
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f2423766db0>, batch_size=1, drop_last=False
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/is2re/val_20/data.lmdb')]'
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d56e4b0>, batch_size=1, drop_last=False
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy'] have been saved to: ./checkpoints/2025-03-29-04-45-52-IS2RE-example/normalizers.pt
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy', 'forces'] have been saved to: ./checkpoints/2025-03-29-04-45-52-IS2RE-example/normalizers.pt
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output energy: mean=-1.4570415561499996, rmsd=1.8371084209427546.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output forces: mean=0, rmsd=1.
Train the Model#
energy_trainer.train()
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_0_sum (out_blocks.0.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_1_sum (out_blocks.1.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_2_sum (out_blocks.2.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_3_sum (out_blocks.3.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_4_sum (out_blocks.4.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_5_sum (out_blocks.5.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_1_had_rbf (int_blocks.0.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_1_sum_cbf (int_blocks.0.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_1_sum (int_blocks.0.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_2_had_rbf (int_blocks.1.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_2_sum_cbf (int_blocks.1.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_2_sum (int_blocks.1.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_3_had_rbf (int_blocks.2.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_3_sum_cbf (int_blocks.2.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_3_sum (int_blocks.2.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_4_had_rbf (int_blocks.3.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_4_sum_cbf (int_blocks.3.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_4_sum (int_blocks.3.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_5_had_rbf (int_blocks.4.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_5_sum_cbf (int_blocks.4.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_5_sum (int_blocks.4.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:45:44 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 4.10e+01, energy_mse: 3.12e+03, energy_within_threshold: 0.00e+00, loss: 2.23e+01, lr: 1.00e-04, epoch: 5.00e-02, step: 5.00e+00
2025-03-29 04:45:55 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.19e+02, energy_mse: 2.32e+04, energy_within_threshold: 0.00e+00, loss: 6.47e+01, lr: 1.00e-04, epoch: 1.00e-01, step: 1.00e+01
2025-03-29 04:46:09 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.64e+02, energy_mse: 4.57e+04, energy_within_threshold: 0.00e+00, loss: 8.95e+01, lr: 1.00e-04, epoch: 1.50e-01, step: 1.50e+01
2025-03-29 04:46:21 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.28e+02, energy_mse: 2.71e+04, energy_within_threshold: 0.00e+00, loss: 6.96e+01, lr: 1.00e-04, epoch: 2.00e-01, step: 2.00e+01
2025-03-29 04:46:34 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 2.05e+02, energy_mse: 4.59e+04, energy_within_threshold: 0.00e+00, loss: 1.12e+02, lr: 1.00e-04, epoch: 2.50e-01, step: 2.50e+01
2025-03-29 04:46:44 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 9.08e+01, energy_mse: 1.17e+04, energy_within_threshold: 0.00e+00, loss: 4.95e+01, lr: 1.00e-04, epoch: 3.00e-01, step: 3.00e+01
2025-03-29 04:46:59 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 2.48e+02, energy_mse: 9.34e+04, energy_within_threshold: 0.00e+00, loss: 1.35e+02, lr: 1.00e-04, epoch: 3.50e-01, step: 3.50e+01
2025-03-29 04:47:08 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 9.97e+01, energy_mse: 1.46e+04, energy_within_threshold: 0.00e+00, loss: 5.43e+01, lr: 1.00e-04, epoch: 4.00e-01, step: 4.00e+01
2025-03-29 04:47:21 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.54e+02, energy_mse: 3.14e+04, energy_within_threshold: 0.00e+00, loss: 8.38e+01, lr: 1.00e-04, epoch: 4.50e-01, step: 4.50e+01
2025-03-29 04:47:39 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.65e+02, energy_mse: 4.05e+04, energy_within_threshold: 0.00e+00, loss: 8.98e+01, lr: 1.00e-04, epoch: 5.00e-01, step: 5.00e+01
2025-03-29 04:47:48 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.25e+02, energy_mse: 3.25e+04, energy_within_threshold: 0.00e+00, loss: 6.78e+01, lr: 1.00e-04, epoch: 5.50e-01, step: 5.50e+01
2025-03-29 04:48:00 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.36e+02, energy_mse: 3.34e+04, energy_within_threshold: 0.00e+00, loss: 7.41e+01, lr: 1.00e-04, epoch: 6.00e-01, step: 6.00e+01
2025-03-29 04:48:11 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.03e+02, energy_mse: 1.22e+04, energy_within_threshold: 0.00e+00, loss: 5.61e+01, lr: 1.00e-04, epoch: 6.50e-01, step: 6.50e+01
2025-03-29 04:48:21 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.35e+02, energy_mse: 4.46e+04, energy_within_threshold: 0.00e+00, loss: 7.36e+01, lr: 1.00e-04, epoch: 7.00e-01, step: 7.00e+01
2025-03-29 04:48:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.73e+02, energy_mse: 4.67e+04, energy_within_threshold: 0.00e+00, loss: 9.41e+01, lr: 1.00e-04, epoch: 7.50e-01, step: 7.50e+01
2025-03-29 04:48:43 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 9.48e+01, energy_mse: 1.10e+04, energy_within_threshold: 0.00e+00, loss: 5.16e+01, lr: 1.00e-04, epoch: 8.00e-01, step: 8.00e+01
2025-03-29 04:48:57 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 5.78e+01, energy_mse: 5.72e+03, energy_within_threshold: 0.00e+00, loss: 3.15e+01, lr: 1.00e-04, epoch: 8.50e-01, step: 8.50e+01
2025-03-29 04:49:07 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.34e+02, energy_mse: 6.19e+04, energy_within_threshold: 0.00e+00, loss: 7.28e+01, lr: 1.00e-04, epoch: 9.00e-01, step: 9.00e+01
2025-03-29 04:49:20 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 1.23e+02, energy_mse: 1.90e+04, energy_within_threshold: 0.00e+00, loss: 6.71e+01, lr: 1.00e-04, epoch: 9.50e-01, step: 9.50e+01
2025-03-29 04:49:29 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_mae: 8.00e+01, energy_mse: 1.03e+04, energy_within_threshold: 0.00e+00, loss: 4.36e+01, lr: 1.00e-04, epoch: 1.00e+00, step: 1.00e+02
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_0_sum (out_blocks.0.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_1_sum (out_blocks.1.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_2_sum (out_blocks.2.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_3_sum (out_blocks.3.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_4_sum (out_blocks.4.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_5_sum (out_blocks.5.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_1_had_rbf (int_blocks.0.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_1_sum_cbf (int_blocks.0.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_1_sum (int_blocks.0.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_2_had_rbf (int_blocks.1.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_2_sum_cbf (int_blocks.1.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_2_sum (int_blocks.1.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_3_had_rbf (int_blocks.2.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_3_sum_cbf (int_blocks.2.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_3_sum (int_blocks.2.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_4_had_rbf (int_blocks.3.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_4_sum_cbf (int_blocks.3.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_4_sum (int_blocks.3.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_5_had_rbf (int_blocks.4.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_5_sum_cbf (int_blocks.4.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_5_sum (int_blocks.4.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/20 [00:00<?, ?it/s]
device 0: 5%|▌ | 1/20 [00:01<00:21, 1.15s/it]
device 0: 10%|█ | 2/20 [00:02<00:21, 1.17s/it]
device 0: 15%|█▌ | 3/20 [00:03<00:20, 1.20s/it]
device 0: 20%|██ | 4/20 [00:04<00:17, 1.09s/it]
device 0: 25%|██▌ | 5/20 [00:05<00:14, 1.05it/s]
device 0: 30%|███ | 6/20 [00:06<00:14, 1.06s/it]
device 0: 35%|███▌ | 7/20 [00:07<00:14, 1.14s/it]
device 0: 40%|████ | 8/20 [00:08<00:11, 1.08it/s]
device 0: 45%|████▌ | 9/20 [00:09<00:10, 1.08it/s]
device 0: 50%|█████ | 10/20 [00:11<00:14, 1.48s/it]
device 0: 55%|█████▌ | 11/20 [00:12<00:11, 1.33s/it]
device 0: 60%|██████ | 12/20 [00:13<00:09, 1.17s/it]
device 0: 65%|██████▌ | 13/20 [00:14<00:08, 1.14s/it]
device 0: 70%|███████ | 14/20 [00:15<00:05, 1.08it/s]
device 0: 75%|███████▌ | 15/20 [00:15<00:03, 1.48it/s]
device 0: 80%|████████ | 16/20 [00:15<00:02, 1.63it/s]
device 0: 85%|████████▌ | 17/20 [00:16<00:01, 1.68it/s]
device 0: 90%|█████████ | 18/20 [00:17<00:01, 1.28it/s]
device 0: 95%|█████████▌| 19/20 [00:17<00:00, 1.48it/s]
device 0: 100%|██████████| 20/20 [00:18<00:00, 1.53it/s]
device 0: 100%|██████████| 20/20 [00:18<00:00, 1.07it/s]
2025-03-29 04:49:48 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_mae: 56.5138, energy_mse: 6687.5212, energy_within_threshold: 0.0000, loss: 30.7624, epoch: 1.0000
Validate the Model#
Load the best checkpoint#
# The `best_checpoint.pt` file contains the checkpoint with the best val performance
checkpoint_path = os.path.join(energy_trainer.config["cmd"]["checkpoint_dir"], "best_checkpoint.pt")
checkpoint_path
'./checkpoints/2025-03-29-04-45-52-IS2RE-example/best_checkpoint.pt'
# Append the dataset with the test set. We use the same val set for demonstration.
# Dataset
dataset.append(
{'src': val_src}, # test set (optional)
)
dataset
[{'src': 'data/is2re/train_100/data.lmdb',
'normalize_labels': True,
'target_mean': -1.4570415561499996,
'target_std': 1.8371084209427546},
{'src': 'data/is2re/val_20/data.lmdb'},
{'src': 'data/is2re/val_20/data.lmdb'}]
pretrained_energy_trainer = OCPTrainer(
task=task,
model=copy.deepcopy(model), # copied for later use, not necessary in practice.
dataset=dataset,
optimizer=optimizer,
outputs={},
loss_functions={},
evaluation_metrics={},
name="is2re",
identifier="IS2RE-val-example",
run_dir="./", # directory to save results if is_debug=False. Prediction files are saved here so be careful not to override!
is_debug=False, # if True, do not save checkpoint, logs, or results
print_every=5,
seed=0, # random seed to use
logger="tensorboard", # logger of choice (tensorboard and wandb supported)
local_rank=0,
amp=True, # use PyTorch Automatic Mixed Precision (faster training and less memory usage),
)
pretrained_energy_trainer.load_checkpoint(checkpoint_path=checkpoint_path)
2025-03-29 04:49:49 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:1319: (WARNING): Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
2025-03-29 04:49:49 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:208: (INFO): amp: true
cmd:
checkpoint_dir: ./checkpoints/2025-03-29-04-50-08-IS2RE-val-example
commit: core:b88b66e,experimental:NA
identifier: IS2RE-val-example
logs_dir: ./logs/tensorboard/2025-03-29-04-50-08-IS2RE-val-example
print_every: 5
results_dir: ./results/2025-03-29-04-50-08-IS2RE-val-example
seed: 0
timestamp_id: 2025-03-29-04-50-08-IS2RE-val-example
version: 0.1.dev1+gb88b66e
dataset:
format: single_point_lmdb
key_mapping:
y_relaxed: energy
normalize_labels: true
src: data/is2re/train_100/data.lmdb
target_mean: !!python/object/apply:numpy.core.multiarray.scalar
- &id001 !!python/object/apply:numpy.dtype
args:
- f8
- false
- true
state: !!python/tuple
- 3
- <
- null
- null
- null
- -1
- -1
- 0
- !!binary |
MjyJzgpQ978=
target_std: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
PnyyzMtk/T8=
transforms:
normalizer:
energy:
mean: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
MjyJzgpQ978=
stdev: !!python/object/apply:numpy.core.multiarray.scalar
- *id001
- !!binary |
PnyyzMtk/T8=
forces:
mean: 0
stdev: 1
evaluation_metrics:
metrics:
energy:
- mae
- mse
- energy_within_threshold
gp_gpus: null
gpus: 0
logger: tensorboard
loss_functions:
- energy:
coefficient: 1
fn: mae
model:
activation: silu
cbf:
name: spherical_harmonics
cutoff: 6.0
direct_forces: false
emb_size_atom: 256
emb_size_bil_trip: 64
emb_size_cbf: 16
emb_size_edge: 512
emb_size_rbf: 16
emb_size_trip: 64
envelope:
exponent: 5
name: polynomial
extensive: true
max_neighbors: 50
name: gemnet_t
num_after_skip: 2
num_atom: 3
num_before_skip: 1
num_blocks: 5
num_concat: 1
num_radial: 64
num_spherical: 7
otf_graph: false
output_init: HeOrthogonal
rbf:
name: gaussian
regress_forces: false
optim:
batch_size: 1
clip_grad_norm: 10
ema_decay: 0.999
eval_batch_size: 1
factor: 0.8
loss_energy: mae
lr_initial: 0.0001
max_epochs: 1
mode: min
num_workers: 2
optimizer: AdamW
optimizer_params:
amsgrad: true
patience: 3
scheduler: ReduceLROnPlateau
outputs:
energy:
level: system
relax_dataset: {}
slurm: {}
task:
dataset: single_point_lmdb
description: Relaxed state energy prediction from initial structure.
labels:
- relaxed energy
metric: mae
type: regression
test_dataset:
src: data/is2re/val_20/data.lmdb
trainer: is2re
val_dataset:
src: data/is2re/val_20/data.lmdb
2025-03-29 04:49:49 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:557: (INFO): Loading model: gemnet_t
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:568: (INFO): Loaded GemNetT with 22774037 parameters.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/logger.py:175: (WARNING): log_summary for Tensorboard not supported
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:358: (INFO): Loading dataset: single_point_lmdb
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/is2re/train_100/data.lmdb')]'
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d2406b0>, batch_size=1, drop_last=False
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/is2re/val_20/data.lmdb')]'
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d2408f0>, batch_size=1, drop_last=False
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/is2re/val_20/data.lmdb')]'
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d5c4c20>, batch_size=1, drop_last=False
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy'] have been saved to: ./checkpoints/2025-03-29-04-50-08-IS2RE-val-example/normalizers.pt
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy', 'forces'] have been saved to: ./checkpoints/2025-03-29-04-50-08-IS2RE-val-example/normalizers.pt
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output energy: mean=-1.4570415561499996, rmsd=1.8371084209427546.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output forces: mean=0, rmsd=1.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:606: (INFO): Loading checkpoint from: ./checkpoints/2025-03-29-04-45-52-IS2RE-example/best_checkpoint.pt
/home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:607: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
checkpoint = torch.load(checkpoint_path, map_location=map_location)
Test the model#
# make predictions on the existing test_loader
predictions = pretrained_energy_trainer.predict(pretrained_trainer.test_loader, results_file="is2re_results", disable_tqdm=False)
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_0_sum (out_blocks.0.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_1_sum (out_blocks.1.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_2_sum (out_blocks.2.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_3_sum (out_blocks.3.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_4_sum (out_blocks.4.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor OutBlock_5_sum (out_blocks.5.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_1_had_rbf (int_blocks.0.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_1_sum_cbf (int_blocks.0.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_1_sum (int_blocks.0.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_2_had_rbf (int_blocks.1.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_2_sum_cbf (int_blocks.1.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_2_sum (int_blocks.1.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_3_had_rbf (int_blocks.2.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_3_sum_cbf (int_blocks.2.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_3_sum (int_blocks.2.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_4_had_rbf (int_blocks.3.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_4_sum_cbf (int_blocks.3.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_4_sum (int_blocks.3.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_5_had_rbf (int_blocks.4.trip_interaction.scale_rbf) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor TripInteraction_5_sum_cbf (int_blocks.4.trip_interaction.scale_cbf_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/util.py:23: (WARNING): Scale factor AtomUpdate_5_sum (int_blocks.4.atom_update.scale_sum) is not fitted. Please make sure that you either (1) load a checkpoint with fitted scale factors, (2) explicitly load scale factors using the `model.scale_file` attribute, or (3) fit the scale factors using the `fit.py` script.
2025-03-29 04:49:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:445: (INFO): Predicting on test.
device 0: 0%| | 0/20 [00:00<?, ?it/s]
device 0: 5%|▌ | 1/20 [00:00<00:05, 3.53it/s]
device 0: 10%|█ | 2/20 [00:00<00:04, 4.16it/s]
device 0: 15%|█▌ | 3/20 [00:00<00:03, 4.35it/s]
device 0: 20%|██ | 4/20 [00:00<00:03, 4.53it/s]
device 0: 25%|██▌ | 5/20 [00:01<00:03, 4.70it/s]
device 0: 30%|███ | 6/20 [00:01<00:02, 4.78it/s]
device 0: 35%|███▌ | 7/20 [00:01<00:02, 4.72it/s]
device 0: 40%|████ | 8/20 [00:01<00:02, 4.77it/s]
device 0: 45%|████▌ | 9/20 [00:01<00:02, 4.74it/s]
device 0: 50%|█████ | 10/20 [00:02<00:02, 4.82it/s]
device 0: 55%|█████▌ | 11/20 [00:02<00:01, 4.79it/s]
device 0: 60%|██████ | 12/20 [00:02<00:01, 4.82it/s]
device 0: 65%|██████▌ | 13/20 [00:02<00:01, 4.88it/s]
device 0: 70%|███████ | 14/20 [00:02<00:01, 4.89it/s]
device 0: 75%|███████▌ | 15/20 [00:03<00:01, 4.95it/s]
device 0: 80%|████████ | 16/20 [00:03<00:00, 4.89it/s]
device 0: 85%|████████▌ | 17/20 [00:03<00:00, 4.88it/s]
device 0: 90%|█████████ | 18/20 [00:03<00:00, 4.83it/s]
device 0: 95%|█████████▌| 19/20 [00:03<00:00, 4.93it/s]
device 0: 100%|██████████| 20/20 [00:04<00:00, 4.94it/s]
device 0: 100%|██████████| 20/20 [00:04<00:00, 4.73it/s]
2025-03-29 04:49:54 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:1006: (INFO): Writing results to ./results/2025-03-29-04-50-08-IS2RE-val-example/is2re_is2re_results.npz
energies = predictions["energy"]
Initial Structure to Relaxed Structure (IS2RS) #
We approach the IS2RS task by using a pre-trained S2EF model to iteratively run a structure optimization to arrive at a relaxed structure. While the majority of approaches for this task do this iteratively, we note it’s possible to train a model to directly predict relaxed structures.
Steps for making IS2RS predictions#
Define or load a configuration (config), which includes the following
task with relaxation dataset information
model
optimizer
dataset
trainer
Create a ForcesTrainer object
Train a S2EF model or load an existing S2EF checkpoint
Run relaxations
Note For this task we’ll be using a publicly released pre-trained checkpoint of our best model to perform relaxations.
Imports#
from fairchem.core.trainers import OCPTrainer
from fairchem.core.datasets import LmdbDataset
from fairchem.core import models
from fairchem.core.common import logger
from fairchem.core.common.utils import setup_logging
setup_logging()
import numpy as np
Dataset#
The IS2RS task requires an additional realxation dataset to be defined - relax_dataset. This dataset is read in similar to the IS2RE dataset - requiring an LMDB file. The same datasets are used for the IS2RE and IS2RS tasks.
train_src = "data/s2ef/train_100"
val_src = "data/s2ef/val_20"
relax_dataset = "data/is2re/val_20/data.lmdb"
Download pretrained checkpoint#
from fairchem.core.models.model_registry import model_name_to_local_file
checkpoint_path = model_name_to_local_file('GemNet-dT-S2EF-OC20-All', local_cache='/tmp/fairchem_checkpoints/')
2025-03-29 04:49:54 /home/runner/work/fairchem/fairchem/src/fairchem/core/models/model_registry.py:59: (INFO): Checking local cache: /tmp/fairchem_checkpoints/ for model GemNet-dT-S2EF-OC20-All
Define the Config#
Running an iterative S2EF model for the IS2RS task can be run from any S2EF config given the following additions to the task portion of the config:
relax_dataset - IS2RE LMDB dataset
write_pos - Whether to save out relaxed positions
relaxation_steps - Number of optimization steps to run
relax_opt - Dictionary of optimizer settings. Currently only LBFGS supported
maxstep - Maximum distance an optimization is allowed to make
memory - Memory history to use for LBFGS
damping - Calculated step is multiplied by this factor before updating positions
alpha - Initial guess for the Hessian
traj_dir - If specified, directory to save out the full ML relaxation as an ASE trajectory. Useful for debugging or visualizing results.
num_relaxation_batches - If specified, relaxations will only be run for a subset of the relaxation dataset. Useful for debugging or wanting to visualize a few systems.
A sample relaxation config can be found here.
# Task
task = {
'dataset': 'lmdb', # dataset used for the S2EF task
'description': 'Regressing to energies and forces for DFT trajectories from OCP',
'type': 'regression',
'metric': 'mae',
'labels': ['potential energy'],
'grad_input': 'atomic forces',
'train_on_free_atoms': True,
'eval_on_free_atoms': True,
'write_pos': True,
'relaxation_steps': 200,
'num_relaxation_batches': 1,
'relax_opt': {
'maxstep': 0.04,
'memory': 50,
'damping': 1.0,
'alpha': 70.0,
'traj_dir': "ml-relaxations/is2rs-test",
}
}
# Model
model = {
'name': 'gemnet_t',
"num_spherical": 7,
"num_radial": 128,
"num_blocks": 3,
"emb_size_atom": 512,
"emb_size_edge": 512,
"emb_size_trip": 64,
"emb_size_rbf": 16,
"emb_size_cbf": 16,
"emb_size_bil_trip": 64,
"num_before_skip": 1,
"num_after_skip": 2,
"num_concat": 1,
"num_atom": 3,
"cutoff": 6.0,
"max_neighbors": 50,
"rbf": {"name": "gaussian"},
"envelope": {
"name": "polynomial",
"exponent": 5,
},
"cbf": {"name": "spherical_harmonics"},
"extensive": True,
"otf_graph": False,
"output_init": "HeOrthogonal",
"activation": "silu",
"scale_file": "./gemnet-dT.json",
"regress_forces": True,
"direct_forces": True,
}
# Optimizer
optimizer = {
'batch_size': 1, # originally 32
'eval_batch_size': 1, # originally 32
'num_workers': 2,
'lr_initial': 5.e-4,
'optimizer': 'AdamW',
'optimizer_params': {"amsgrad": True},
'scheduler': "ReduceLROnPlateau",
'mode': "min",
'factor': 0.8,
'ema_decay': 0.999,
'clip_grad_norm': 10,
'patience': 3,
'max_epochs': 1, # used for demonstration purposes
'force_coefficient': 100,
}
# Dataset
dataset = {
'train': {'src': train_src, 'normalize_labels': False}, # train set
'val': {'src': val_src}, # val set (optional)
'relax': {"src": relax_dataset},
}
Create the trainer#
trainer = OCPTrainer(
task=task,
model=copy.deepcopy(model), # copied for later use, not necessary in practice.
dataset=dataset,
optimizer=optimizer,
outputs={},
loss_functions={},
evaluation_metrics={},
name="s2ef",
identifier="is2rs-example",
run_dir="./", # directory to save results if is_debug=False. Prediction files are saved here so be careful not to override!
is_debug=False, # if True, do not save checkpoint, logs, or results
print_every=5,
seed=0, # random seed to use
logger="tensorboard", # logger of choice (tensorboard and wandb supported)
local_rank=0,
amp=True, # use PyTorch Automatic Mixed Precision (faster training and less memory usage),
)
2025-03-29 04:49:55 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:1319: (WARNING): Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
2025-03-29 04:49:55 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:208: (INFO): amp: true
cmd:
checkpoint_dir: ./checkpoints/2025-03-29-04-50-08-is2rs-example
commit: core:b88b66e,experimental:NA
identifier: is2rs-example
logs_dir: ./logs/tensorboard/2025-03-29-04-50-08-is2rs-example
print_every: 5
results_dir: ./results/2025-03-29-04-50-08-is2rs-example
seed: 0
timestamp_id: 2025-03-29-04-50-08-is2rs-example
version: 0.1.dev1+gb88b66e
dataset:
format: lmdb
key_mapping:
force: forces
y: energy
normalize_labels: false
src: data/s2ef/train_100
evaluation_metrics:
metrics:
energy:
- mae
forces:
- forcesx_mae
- forcesy_mae
- forcesz_mae
- mae
- cosine_similarity
- magnitude_error
misc:
- energy_forces_within_threshold
gp_gpus: null
gpus: 0
logger: tensorboard
loss_functions:
- energy:
coefficient: 1
fn: mae
- forces:
coefficient: 100
fn: l2mae
model:
activation: silu
cbf:
name: spherical_harmonics
cutoff: 6.0
direct_forces: true
emb_size_atom: 512
emb_size_bil_trip: 64
emb_size_cbf: 16
emb_size_edge: 512
emb_size_rbf: 16
emb_size_trip: 64
envelope:
exponent: 5
name: polynomial
extensive: true
max_neighbors: 50
name: gemnet_t
num_after_skip: 2
num_atom: 3
num_before_skip: 1
num_blocks: 3
num_concat: 1
num_radial: 128
num_spherical: 7
otf_graph: false
output_init: HeOrthogonal
rbf:
name: gaussian
regress_forces: true
scale_file: ./gemnet-dT.json
optim:
batch_size: 1
clip_grad_norm: 10
ema_decay: 0.999
eval_batch_size: 1
factor: 0.8
force_coefficient: 100
lr_initial: 0.0005
max_epochs: 1
mode: min
num_workers: 2
optimizer: AdamW
optimizer_params:
amsgrad: true
patience: 3
scheduler: ReduceLROnPlateau
outputs:
energy:
level: system
forces:
eval_on_free_atoms: true
level: atom
train_on_free_atoms: true
relax_dataset:
src: data/is2re/val_20/data.lmdb
slurm: {}
task:
dataset: lmdb
description: Regressing to energies and forces for DFT trajectories from OCP
eval_on_free_atoms: true
grad_input: atomic forces
labels:
- potential energy
metric: mae
num_relaxation_batches: 1
relax_opt:
alpha: 70.0
damping: 1.0
maxstep: 0.04
memory: 50
traj_dir: ml-relaxations/is2rs-test
relaxation_steps: 200
train_on_free_atoms: true
type: regression
write_pos: true
test_dataset: {}
trainer: s2ef
val_dataset:
src: data/s2ef/val_20
2025-03-29 04:49:55 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:557: (INFO): Loading model: gemnet_t
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:568: (INFO): Loaded GemNetT with 31671825 parameters.
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/logger.py:175: (WARNING): log_summary for Tensorboard not supported
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:358: (INFO): Loading dataset: lmdb
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/train_100')]'
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d242210>, batch_size=1, drop_last=False
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/val_20')]'
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245cfe23c0>, batch_size=1, drop_last=False
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/is2re/val_20/data.lmdb')]'
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d242630>, batch_size=1, drop_last=False
Load the best checkpoint#
trainer.load_checkpoint(checkpoint_path=checkpoint_path)
2025-03-29 04:49:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:606: (INFO): Loading checkpoint from: /tmp/fairchem_checkpoints/gemnet_t_direct_h512_all.pt
/home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:607: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
checkpoint = torch.load(checkpoint_path, map_location=map_location)
/home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/normalizer.py:69: UserWarning: To copy construct from a tensor, it is recommended to use sourceTensor.clone().detach() or sourceTensor.clone().detach().requires_grad_(True), rather than torch.tensor(sourceTensor).
"mean": torch.tensor(state_dict["mean"]),
Run relaxations#
We run a full relaxation for a single batch of our relaxation dataset (num_relaxation_batches=1).
trainer.run_relaxations()
2025-03-29 04:49:57 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:567: (INFO): Running ML-relaxations
0%| | 0/20 [00:00<?, ?it/s]
2025-03-29 04:49:58 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:91: (INFO): Step Fmax(eV/A)
2025-03-29 04:49:58 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 0 7.594
/home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/ase_utils.py:105: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calc)
2025-03-29 04:49:59 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 1 5.940
2025-03-29 04:50:00 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 2 4.512
2025-03-29 04:50:01 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 3 3.016
2025-03-29 04:50:02 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 4 3.472
2025-03-29 04:50:03 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 5 3.896
2025-03-29 04:50:04 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 6 4.195
2025-03-29 04:50:05 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 7 4.338
2025-03-29 04:50:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 8 4.322
2025-03-29 04:50:07 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 9 4.156
2025-03-29 04:50:08 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 10 3.865
2025-03-29 04:50:09 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 11 3.461
2025-03-29 04:50:10 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 12 2.972
2025-03-29 04:50:11 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 13 2.432
2025-03-29 04:50:12 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 14 1.880
2025-03-29 04:50:13 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 15 1.365
2025-03-29 04:50:14 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 16 1.185
2025-03-29 04:50:15 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 17 1.125
2025-03-29 04:50:17 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 18 1.010
2025-03-29 04:50:18 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 19 1.084
2025-03-29 04:50:19 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 20 1.156
2025-03-29 04:50:20 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 21 1.198
2025-03-29 04:50:21 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 22 1.215
2025-03-29 04:50:22 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 23 1.208
2025-03-29 04:50:23 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 24 1.177
2025-03-29 04:50:24 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 25 1.128
2025-03-29 04:50:25 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 26 1.069
2025-03-29 04:50:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 27 1.004
2025-03-29 04:50:27 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 28 0.940
2025-03-29 04:50:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 29 0.879
2025-03-29 04:50:29 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 30 0.823
2025-03-29 04:50:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 31 0.787
2025-03-29 04:50:31 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 32 0.816
2025-03-29 04:50:32 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 33 0.842
2025-03-29 04:50:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 34 0.866
2025-03-29 04:50:35 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 35 0.888
2025-03-29 04:50:36 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 36 0.906
2025-03-29 04:50:37 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 37 0.920
2025-03-29 04:50:38 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 38 0.930
2025-03-29 04:50:39 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 39 0.935
2025-03-29 04:50:40 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 40 0.935
2025-03-29 04:50:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 41 0.930
2025-03-29 04:50:42 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 42 0.921
2025-03-29 04:50:43 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 43 0.907
2025-03-29 04:50:44 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 44 0.888
2025-03-29 04:50:45 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 45 0.865
2025-03-29 04:50:46 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 46 0.837
2025-03-29 04:50:47 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 47 0.804
2025-03-29 04:50:48 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 48 0.765
2025-03-29 04:50:49 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 49 0.721
2025-03-29 04:50:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 50 0.672
2025-03-29 04:50:51 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 51 0.619
2025-03-29 04:50:52 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 52 0.564
2025-03-29 04:50:53 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 53 0.508
2025-03-29 04:50:55 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 54 0.466
2025-03-29 04:50:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 55 0.453
2025-03-29 04:50:57 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 56 0.429
2025-03-29 04:50:58 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 57 0.395
2025-03-29 04:50:59 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 58 0.348
2025-03-29 04:51:00 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 59 0.289
2025-03-29 04:51:01 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 60 0.245
2025-03-29 04:51:02 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 61 0.253
2025-03-29 04:51:03 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 62 0.267
2025-03-29 04:51:04 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 63 0.278
2025-03-29 04:51:05 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 64 0.283
2025-03-29 04:51:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 65 0.307
2025-03-29 04:51:07 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 66 0.343
2025-03-29 04:51:08 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 67 0.367
2025-03-29 04:51:09 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 68 0.380
2025-03-29 04:51:10 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 69 0.380
2025-03-29 04:51:11 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 70 0.367
2025-03-29 04:51:12 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 71 0.344
2025-03-29 04:51:14 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 72 0.297
2025-03-29 04:51:15 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 73 0.251
2025-03-29 04:51:16 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 74 0.279
2025-03-29 04:51:17 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 75 0.369
2025-03-29 04:51:18 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 76 0.311
2025-03-29 04:51:19 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 77 0.338
2025-03-29 04:51:20 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 78 0.285
2025-03-29 04:51:21 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 79 0.202
2025-03-29 04:51:22 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 80 0.174
2025-03-29 04:51:23 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 81 0.168
2025-03-29 04:51:24 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 82 0.150
2025-03-29 04:51:25 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 83 0.147
2025-03-29 04:51:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 84 0.140
2025-03-29 04:51:27 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 85 0.138
2025-03-29 04:51:28 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 86 0.150
2025-03-29 04:51:29 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 87 0.165
2025-03-29 04:51:30 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 88 0.135
2025-03-29 04:51:31 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 89 0.106
2025-03-29 04:51:33 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 90 0.087
2025-03-29 04:51:34 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 91 0.092
2025-03-29 04:51:35 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 92 0.103
2025-03-29 04:51:36 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 93 0.075
2025-03-29 04:51:37 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 94 0.054
2025-03-29 04:51:38 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 95 0.034
2025-03-29 04:51:39 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 96 0.028
2025-03-29 04:51:40 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 97 0.028
2025-03-29 04:51:41 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 98 0.039
2025-03-29 04:51:42 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 99 0.076
2025-03-29 04:51:43 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 100 0.052
2025-03-29 04:51:44 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 101 0.029
2025-03-29 04:51:45 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 102 0.041
2025-03-29 04:51:46 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 103 0.052
2025-03-29 04:51:47 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 104 0.032
2025-03-29 04:51:48 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 105 0.031
2025-03-29 04:51:49 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 106 0.061
2025-03-29 04:51:50 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 107 0.048
2025-03-29 04:51:51 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 108 0.024
2025-03-29 04:51:53 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 109 0.028
2025-03-29 04:51:54 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 110 0.023
2025-03-29 04:51:55 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 111 0.032
2025-03-29 04:51:56 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 112 0.040
2025-03-29 04:51:57 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 113 0.057
2025-03-29 04:51:58 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 114 0.026
2025-03-29 04:51:59 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 115 0.024
2025-03-29 04:52:00 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 116 0.024
2025-03-29 04:52:01 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 117 0.024
2025-03-29 04:52:02 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:96: (INFO): 118 0.024
2025-03-29 04:52:03 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/optimizers/lbfgs_torch.py:109: (INFO): 119 0.019
5%|▌ | 1/20 [02:06<40:02, 126.46s/it]
5%|▌ | 1/20 [02:06<40:03, 126.48s/it]
2025-03-29 04:52:03 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:694: (INFO): Writing results to ./results/2025-03-29-04-50-08-is2rs-example/relaxed_positions.npz
Visualize ML-driven relaxations#
Following our earlier visualization steps, we can plot our ML-generated relaxations.
import glob
import ase.io
from ase.visualize.plot import plot_atoms
import matplotlib.pyplot as plt
import random
import matplotlib
params = {
'axes.labelsize': 14,
'font.size': 14,
'font.family': ' DejaVu Sans',
'legend.fontsize': 20,
'xtick.labelsize': 20,
'ytick.labelsize': 20,
'axes.labelsize': 25,
'axes.titlesize': 25,
'text.usetex': False,
'figure.figsize': [12, 12]
}
matplotlib.rcParams.update(params)
system = glob.glob("ml-relaxations/is2rs-test/*.traj")[0]
ml_trajectory = ase.io.read(system, ":")
energies = [atom.get_potential_energy() for atom in ml_trajectory]
plt.figure(figsize=(7, 5))
plt.plot(range(len(energies)), energies)
plt.xlabel("step")
plt.ylabel("energy, eV")
system
'ml-relaxations/is2rs-test/1700380.traj'
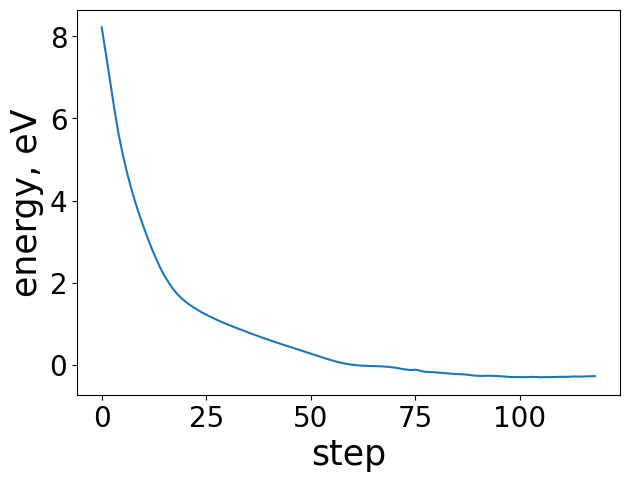
Qualitatively, the ML relaxation is behaving as expected - decreasing energies over the course of the relaxation.
fig, ax = plt.subplots(1, 3)
labels = ['ml-initial', 'ml-middle', 'ml-final']
for i in range(3):
ax[i].axis('off')
ax[i].set_title(labels[i])
ase.visualize.plot.plot_atoms(
ml_trajectory[0],
ax[0],
radii=0.8,
# rotation=("-75x, 45y, 10z")) # uncomment to visualize at different angles
)
ase.visualize.plot.plot_atoms(
ml_trajectory[100],
ax[1],
radii=0.8,
# rotation=("-75x, 45y, 10z") # uncomment to visualize at different angles
)
ase.visualize.plot.plot_atoms(
ml_trajectory[-1],
ax[2],
radii=0.8,
# rotation=("-75x, 45y, 10z"), # uncomment to visualize at different angles
)
<Axes: title={'center': 'ml-final'}>
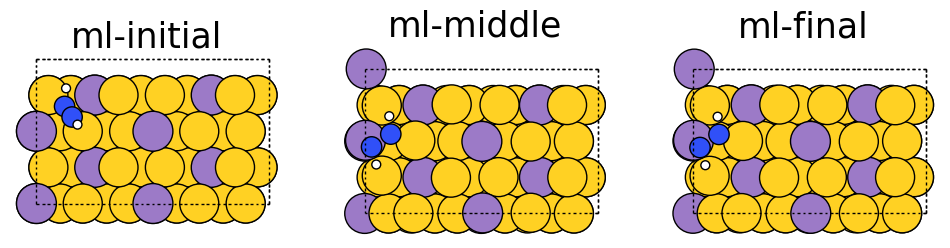
Qualitatively, the generated structures seem reasonable with no obvious issues we had previously mentioned to look out for.
Model development #
In this section, we will walk through how to develop a simple Graph Neural Network model on the S2EF-200k dataset.
Let’s begin by setting up some imports and boilerplate config parameters.
Imports#
import torch
from typing import Optional
from fairchem.core.trainers import OCPTrainer
from fairchem.core import models
from fairchem.core.common import logger
from fairchem.core.common.utils import setup_logging, get_pbc_distances
from fairchem.core.common.registry import registry
from fairchem.core.models.gemnet.layers.radial_basis import PolynomialEnvelope
from torch_geometric.nn.models.schnet import GaussianSmearing
from torch_scatter import scatter
setup_logging()
# Dataset paths
train_src = "data/s2ef/train_100"
val_src = "data/s2ef/val_20"
# Configs
task = {
'dataset': 'trajectory_lmdb', # dataset used for the S2EF task
'description': 'Regressing to energies and forces for DFT trajectories from OCP',
'type': 'regression',
'metric': 'mae',
'labels': ['potential energy'],
'grad_input': 'atomic forces',
'train_on_free_atoms': True,
'eval_on_free_atoms': True
}
# Optimizer
optimizer = {
'batch_size': 16, # if hitting GPU memory issues, lower this
'eval_batch_size': 8,
'num_workers': 8,
'lr_initial': 0.0001,
'scheduler': "ReduceLROnPlateau",
'mode': "min",
'factor': 0.8,
'patience': 3,
'max_epochs': 80,
'max_epochs': 5,
'force_coefficient': 100,
}
# Dataset
dataset = [
{'src': train_src, 'normalize_labels': True, 'target_mean': -0.7554450631141663, 'target_std': 2.887317180633545, 'grad_target_mean': 0.0, 'grad_target_std': 2.887317180633545}, # train set
{'src': val_src},
]
Atom and Edge Embeddings#
Each atom is represented as a node with its features computed using a simple torch.nn.Embedding layer on the atomic number.
All pairs of atoms with a defined cutoff radius (=6A) are assumed to have edges between them, with their features computed as the concatenation of 1) a Gaussian expansion of the distance between the atoms, and the 2) source and 3) target node features.
We will use the GaussianSmearing layer (reproduced below) from the PyTorch Geometric library for computing distance features:
class GaussianSmearing(torch.nn.Module):
def __init__(self, start=0.0, stop=5.0, num_gaussians=50):
super(GaussianSmearing, self).__init__()
offset = torch.linspace(start, stop, num_gaussians)
self.coeff = -0.5 / (offset[1] - offset[0]).item()**2
self.register_buffer('offset', offset)
def forward(self, dist):
dist = dist.view(-1, 1) - self.offset.view(1, -1)
return torch.exp(self.coeff * torch.pow(dist, 2))
class AtomEmbedding(torch.nn.Module):
def __init__(self, emb_size):
super().__init__()
self.embeddings = torch.nn.Embedding(83, emb_size) # We go up to Bi (83).
def forward(self, Z):
h = self.embeddings(Z - 1) # -1 because Z.min()=1 (==Hydrogen)
return h
class EdgeEmbedding(torch.nn.Module):
def __init__(self, atom_emb_size, edge_emb_size, out_size):
super().__init__()
in_features = 2 * atom_emb_size + edge_emb_size
self.dense = torch.nn.Sequential(
torch.nn.Linear(in_features, out_size, bias=False),
torch.nn.SiLU()
)
def forward(self, h, m_rbf, idx_s, idx_t,
):
h_s = h[idx_s] # indexing source node, shape=(num_edges, emb_size)
h_t = h[idx_t] # indexing target node, shape=(num_edges, emb_size)
m_st = torch.cat([h_s, h_t, m_rbf], dim=-1) # (num_edges, 2 * atom_emb_size + edge_emb_size)
m_st = self.dense(m_st) # (num_edges, out_size)
return m_st
class RadialBasis(torch.nn.Module):
def __init__(self, num_radial: int, cutoff: float, env_exponent: int = 5):
super().__init__()
self.inv_cutoff = 1 / cutoff
self.envelope = PolynomialEnvelope(env_exponent)
self.rbf = GaussianSmearing(start=0, stop=1, num_gaussians=num_radial)
def forward(self, d):
d_scaled = d * self.inv_cutoff
env = self.envelope(d_scaled)
return env[:, None] * self.rbf(d_scaled) # (num_edges, num_radial)
Message passing#
We start by implementing a very simple message-passing scheme to predict system energy and forces.
Given the node and edge features, we sum up edge features for all edges \(e_{ij}\) connecting node \(i\) to its neighbors \(j\), and pass the resultant vector through a fully-connected layer to project it down to a scalar. This gives us a scalar energy contribution for each node \(i\) in the structure. We then sum up all node energy contributions to predict the overall system energy.
Similarly, to predict forces, we pass edge features through a fully-connected layer to project it down to a scalar representing the force magnitude per edge \(e_{ij}\). We can then sum up these force magnitudes based on the original edge directions to predict the resultant force vector per node \(i\).
@registry.register_model("simple")
class SimpleAtomEdgeModel(torch.nn.Module):
def __init__(self, emb_size=64, num_radial=64, cutoff=6.0, env_exponent=5):
super().__init__()
self.radial_basis = RadialBasis(
num_radial=num_radial,
cutoff=cutoff,
env_exponent=env_exponent,
)
self.atom_emb = AtomEmbedding(emb_size)
self.edge_emb = EdgeEmbedding(emb_size, num_radial, emb_size)
self.out_energy = torch.nn.Linear(emb_size, 1)
self.out_forces = torch.nn.Linear(emb_size, 1)
def forward(self, data):
batch = data.batch
atomic_numbers = data.atomic_numbers.long()
edge_index = data.edge_index
cell_offsets = data.cell_offsets
neighbors = data.neighbors
# computing edges and distances taking periodic boundary conditions into account
out = get_pbc_distances(
data.pos,
edge_index,
data.cell,
cell_offsets,
neighbors,
return_offsets=True,
return_distance_vec=True,
)
edge_index = out["edge_index"]
D_st = out["distances"]
V_st = -out["distance_vec"] / D_st[:, None]
idx_s, idx_t = edge_index
# embed atoms
h_atom = self.atom_emb(atomic_numbers)
# gaussian expansion of distances D_st
m_rbf = self.radial_basis(D_st)
# embed edges
m = self.edge_emb(h_atom, m_rbf, idx_s, idx_t)
# read out energy
#
# x_E_i = \sum_j m_ji -- summing up edge features m_ji for all neighbors j
# of node i to predict node i's energy contribution.
x_E = scatter(m, idx_t, dim=0, dim_size=h_atom.shape[0], reduce="sum")
x_E = self.out_energy(x_E)
# E = \sum_i x_E_i
num_systems = torch.max(batch)+1
E = scatter(x_E, batch, dim=0, dim_size=num_systems, reduce="add")
# (num_systems, 1)
# read out forces
#
# x_F is the force magnitude per edge, we multiply that by the direction of each edge ji,
# and sum up all the vectors to predict the resultant force on node i
x_F = self.out_forces(m)
F_st_vec = x_F[:, :, None] * V_st[:, None, :]
F = scatter(F_st_vec, idx_t, dim=0, dim_size=atomic_numbers.size(0), reduce="add")
# (num_atoms, num_targets, 3)
F = F.squeeze(1)
return {"energy": E, "forces": F}
@property
def num_params(self):
return sum(p.numel() for p in self.parameters())
Training the model#
model_params = {
'name': 'simple',
'emb_size': 256,
'num_radial': 128,
'cutoff': 6.0,
'env_exponent': 5,
}
trainer = OCPTrainer(
task=task,
model=model_params,
dataset=dataset,
optimizer=optimizer,
outputs={},
loss_functions={},
evaluation_metrics={},
name="s2ef",
identifier="S2EF-simple",
run_dir="./", # directory to save results if is_debug=False. Prediction files are saved here so be careful not to override!
is_debug=False, # if True, do not save checkpoint, logs, or results
print_every=5,
seed=0, # random seed to use
logger="tensorboard", # logger of choice (tensorboard and wandb supported)
local_rank=0,
amp=True, # use PyTorch Automatic Mixed Precision (faster training and less memory usage),
)
trainer.train()
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:1319: (WARNING): Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:208: (INFO): amp: true
cmd:
checkpoint_dir: ./checkpoints/2025-03-29-04-52-16-S2EF-simple
commit: core:b88b66e,experimental:NA
identifier: S2EF-simple
logs_dir: ./logs/tensorboard/2025-03-29-04-52-16-S2EF-simple
print_every: 5
results_dir: ./results/2025-03-29-04-52-16-S2EF-simple
seed: 0
timestamp_id: 2025-03-29-04-52-16-S2EF-simple
version: 0.1.dev1+gb88b66e
dataset:
format: trajectory_lmdb
grad_target_mean: 0.0
grad_target_std: 2.887317180633545
key_mapping:
force: forces
y: energy
normalize_labels: true
src: data/s2ef/train_100
target_mean: -0.7554450631141663
target_std: 2.887317180633545
transforms:
normalizer:
energy:
mean: -0.7554450631141663
stdev: 2.887317180633545
forces:
mean: 0.0
stdev: 2.887317180633545
evaluation_metrics:
metrics:
energy:
- mae
forces:
- forcesx_mae
- forcesy_mae
- forcesz_mae
- mae
- cosine_similarity
- magnitude_error
misc:
- energy_forces_within_threshold
gp_gpus: null
gpus: 0
logger: tensorboard
loss_functions:
- energy:
coefficient: 1
fn: mae
- forces:
coefficient: 100
fn: l2mae
model:
cutoff: 6.0
emb_size: 256
env_exponent: 5
name: simple
num_radial: 128
optim:
batch_size: 16
eval_batch_size: 8
factor: 0.8
force_coefficient: 100
lr_initial: 0.0001
max_epochs: 5
mode: min
num_workers: 8
patience: 3
scheduler: ReduceLROnPlateau
outputs:
energy:
level: system
forces:
eval_on_free_atoms: true
level: atom
train_on_free_atoms: true
relax_dataset: {}
slurm: {}
task:
dataset: trajectory_lmdb
description: Regressing to energies and forces for DFT trajectories from OCP
eval_on_free_atoms: true
grad_input: atomic forces
labels:
- potential energy
metric: mae
train_on_free_atoms: true
type: regression
test_dataset: {}
trainer: s2ef
val_dataset:
src: data/s2ef/val_20
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:557: (INFO): Loading model: simple
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:568: (INFO): Loaded SimpleAtomEdgeModel with 185602 parameters.
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/logger.py:175: (WARNING): log_summary for Tensorboard not supported
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:358: (INFO): Loading dataset: trajectory_lmdb
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/train_100')]'
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f2440db71a0>, batch_size=16, drop_last=False
/opt/hostedtoolcache/Python/3.12.9/x64/lib/python3.12/site-packages/torch/utils/data/dataloader.py:557: UserWarning: This DataLoader will create 8 worker processes in total. Our suggested max number of worker in current system is 4, which is smaller than what this DataLoader is going to create. Please be aware that excessive worker creation might get DataLoader running slow or even freeze, lower the worker number to avoid potential slowness/freeze if necessary.
warnings.warn(_create_warning_msg(
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/val_20')]'
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f2440db7b30>, batch_size=8, drop_last=False
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy'] have been saved to: ./checkpoints/2025-03-29-04-52-16-S2EF-simple/normalizers.pt
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy', 'forces'] have been saved to: ./checkpoints/2025-03-29-04-52-16-S2EF-simple/normalizers.pt
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output energy: mean=-0.7554450631141663, rmsd=2.887317180633545.
2025-03-29 04:52:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output forces: mean=0.0, rmsd=2.887317180633545.
2025-03-29 04:52:08 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 4.11e+02, forcesx_mae: 3.24e-01, forcesy_mae: 4.73e-01, forcesz_mae: 1.61e+00, forces_mae: 8.01e-01, forces_cosine_similarity: 1.95e-02, forces_magnitude_error: 1.59e+00, loss: 2.07e+02, lr: 1.00e-04, epoch: 7.14e-01, step: 5.00e+00
2025-03-29 04:52:09 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 8.06it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 10.97it/s]
2025-03-29 04:52:10 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 197.5696, forcesx_mae: 0.2953, forcesy_mae: 0.2344, forcesz_mae: 0.8644, forces_mae: 0.4647, forces_cosine_similarity: -0.0500, forces_magnitude_error: 0.7782, loss: 103.0341, epoch: 1.0000
2025-03-29 04:52:12 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 2.80e+02, forcesx_mae: 2.55e-01, forcesy_mae: 3.87e-01, forcesz_mae: 1.02e+00, forces_mae: 5.54e-01, forces_cosine_similarity: -1.38e-02, forces_magnitude_error: 9.78e-01, loss: 1.26e+02, lr: 1.00e-04, epoch: 1.43e+00, step: 1.00e+01
2025-03-29 04:52:13 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 7.91it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 10.71it/s]
2025-03-29 04:52:14 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 191.2872, forcesx_mae: 0.3271, forcesy_mae: 0.2744, forcesz_mae: 0.9364, forces_mae: 0.5126, forces_cosine_similarity: -0.0479, forces_magnitude_error: 0.8843, loss: 104.5027, epoch: 2.0000
2025-03-29 04:52:15 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 2.28e+02, forcesx_mae: 2.67e-01, forcesy_mae: 3.55e-01, forcesz_mae: 1.05e+00, forces_mae: 5.58e-01, forces_cosine_similarity: -1.39e-02, forces_magnitude_error: 1.01e+00, loss: 1.26e+02, lr: 1.00e-04, epoch: 2.14e+00, step: 1.50e+01
2025-03-29 04:52:17 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 2.47e+02, forcesx_mae: 2.23e-01, forcesy_mae: 3.52e-01, forcesz_mae: 8.08e-01, forces_mae: 4.61e-01, forces_cosine_similarity: 7.62e-02, forces_magnitude_error: 7.88e-01, loss: 1.21e+02, lr: 1.00e-04, epoch: 2.86e+00, step: 2.00e+01
2025-03-29 04:52:17 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 7.78it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 10.84it/s]
2025-03-29 04:52:18 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 198.2147, forcesx_mae: 0.3246, forcesy_mae: 0.2725, forcesz_mae: 0.9452, forces_mae: 0.5141, forces_cosine_similarity: -0.0507, forces_magnitude_error: 0.8909, loss: 107.1592, epoch: 3.0000
2025-03-29 04:52:20 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.27e+02, forcesx_mae: 1.63e-01, forcesy_mae: 2.30e-01, forcesz_mae: 4.96e-01, forces_mae: 2.97e-01, forces_cosine_similarity: -3.68e-02, forces_magnitude_error: 4.65e-01, loss: 6.43e+01, lr: 1.00e-04, epoch: 3.57e+00, step: 2.50e+01
2025-03-29 04:52:21 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 8.51it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 10.79it/s]
2025-03-29 04:52:22 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 193.6992, forcesx_mae: 0.3220, forcesy_mae: 0.2708, forcesz_mae: 0.9639, forces_mae: 0.5189, forces_cosine_similarity: -0.0511, forces_magnitude_error: 0.9075, loss: 106.1556, epoch: 4.0000
2025-03-29 04:52:23 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.43e+02, forcesx_mae: 1.99e-01, forcesy_mae: 3.15e-01, forcesz_mae: 5.12e-01, forces_mae: 3.42e-01, forces_cosine_similarity: -1.52e-03, forces_magnitude_error: 5.13e-01, loss: 7.14e+01, lr: 1.00e-04, epoch: 4.29e+00, step: 3.00e+01
2025-03-29 04:52:25 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 2.44e+02, forcesx_mae: 1.45e-01, forcesy_mae: 2.35e-01, forcesz_mae: 3.94e-01, forces_mae: 2.58e-01, forces_cosine_similarity: 3.66e-02, forces_magnitude_error: 4.01e-01, loss: 9.90e+01, lr: 1.00e-04, epoch: 5.00e+00, step: 3.50e+01
2025-03-29 04:52:25 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 67%|██████▋ | 2/3 [00:00<00:00, 13.46it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 10.81it/s]
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 201.6972, forcesx_mae: 0.3385, forcesy_mae: 0.2883, forcesz_mae: 1.0024, forces_mae: 0.5431, forces_cosine_similarity: -0.0537, forces_magnitude_error: 0.9617, loss: 110.8121, epoch: 5.0000
If you’ve wired everything up correctly, this model should be relatively small (~185k params) and achieve a force MAE of 0.0815, force cosine of 0.0321, energy MAE of 2.2772 in 2 epochs.
We encourage the reader to try playing with the embedding size, cutoff radius, number of gaussian basis functions, and polynomial envelope exponent to see how it affects performance.
Incorporating triplets and training GemNet-T#
Recall how this model computes edge embeddings based only on a Gaussian expansion of edge distances.
To better capture 3D geometry, we should also embed angles formed by triplets or quadruplets of atoms. A model that incorporates this idea and works quite well is GemNet (Klicpera et al., NeurIPS 2021); see the following figure.

You can train a GemNet-T (T = triplets) on S2EF-200k using the following config.
Note that this is a significantly bulkier model (~3.4M params) than the one we developed above and will take longer to train.
model_params = {
'name': 'gemnet_t',
'num_spherical': 7,
'num_radial': 128,
'num_blocks': 1,
'emb_size_atom': 256,
'emb_size_edge': 256,
'emb_size_trip': 64,
'emb_size_rbf': 16,
'emb_size_cbf': 16,
'emb_size_bil_trip': 64,
'num_before_skip': 1,
'num_after_skip': 1,
'num_concat': 1,
'num_atom': 3,
'cutoff': 6.0,
'max_neighbors': 50,
'rbf': {'name': 'gaussian'},
'envelope': {'name': 'polynomial', 'exponent': 5},
'cbf': {'name': 'spherical_harmonics'},
'extensive': True,
'otf_graph': False,
'output_init': 'HeOrthogonal',
'activation': 'silu',
'scale_file': './gemnet-dT.json',
'regress_forces': True,
'direct_forces': True,
}
trainer = OCPTrainer(
task=task,
model=model_params,
dataset=dataset,
optimizer=optimizer,
outputs={},
loss_functions={},
evaluation_metrics={},
name="s2ef",
identifier="S2EF-gemnet-t",
run_dir="./", # directory to save results if is_debug=False. Prediction files are saved here so be careful not to override!
is_debug=False, # if True, do not save checkpoint, logs, or results
print_every=5,
seed=0, # random seed to use
logger="tensorboard", # logger of choice (tensorboard and wandb supported)
local_rank=0,
amp=True, # use PyTorch Automatic Mixed Precision (faster training and less memory usage),
)
trainer.train()
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:1319: (WARNING): Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:208: (INFO): amp: true
cmd:
checkpoint_dir: ./checkpoints/2025-03-29-04-52-16-S2EF-gemnet-t
commit: core:b88b66e,experimental:NA
identifier: S2EF-gemnet-t
logs_dir: ./logs/tensorboard/2025-03-29-04-52-16-S2EF-gemnet-t
print_every: 5
results_dir: ./results/2025-03-29-04-52-16-S2EF-gemnet-t
seed: 0
timestamp_id: 2025-03-29-04-52-16-S2EF-gemnet-t
version: 0.1.dev1+gb88b66e
dataset:
format: trajectory_lmdb
grad_target_mean: 0.0
grad_target_std: 2.887317180633545
key_mapping:
force: forces
y: energy
normalize_labels: true
src: data/s2ef/train_100
target_mean: -0.7554450631141663
target_std: 2.887317180633545
transforms:
normalizer:
energy:
mean: -0.7554450631141663
stdev: 2.887317180633545
forces:
mean: 0.0
stdev: 2.887317180633545
evaluation_metrics:
metrics:
energy:
- mae
forces:
- forcesx_mae
- forcesy_mae
- forcesz_mae
- mae
- cosine_similarity
- magnitude_error
misc:
- energy_forces_within_threshold
gp_gpus: null
gpus: 0
logger: tensorboard
loss_functions:
- energy:
coefficient: 1
fn: mae
- forces:
coefficient: 100
fn: l2mae
model:
activation: silu
cbf:
name: spherical_harmonics
cutoff: 6.0
direct_forces: true
emb_size_atom: 256
emb_size_bil_trip: 64
emb_size_cbf: 16
emb_size_edge: 256
emb_size_rbf: 16
emb_size_trip: 64
envelope:
exponent: 5
name: polynomial
extensive: true
max_neighbors: 50
name: gemnet_t
num_after_skip: 1
num_atom: 3
num_before_skip: 1
num_blocks: 1
num_concat: 1
num_radial: 128
num_spherical: 7
otf_graph: false
output_init: HeOrthogonal
rbf:
name: gaussian
regress_forces: true
scale_file: ./gemnet-dT.json
optim:
batch_size: 16
eval_batch_size: 8
factor: 0.8
force_coefficient: 100
lr_initial: 0.0001
max_epochs: 5
mode: min
num_workers: 8
patience: 3
scheduler: ReduceLROnPlateau
outputs:
energy:
level: system
forces:
eval_on_free_atoms: true
level: atom
train_on_free_atoms: true
relax_dataset: {}
slurm: {}
task:
dataset: trajectory_lmdb
description: Regressing to energies and forces for DFT trajectories from OCP
eval_on_free_atoms: true
grad_input: atomic forces
labels:
- potential energy
metric: mae
train_on_free_atoms: true
type: regression
test_dataset: {}
trainer: s2ef
val_dataset:
src: data/s2ef/val_20
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:557: (INFO): Loading model: gemnet_t
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor TripInteraction_2_had_rbf not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor TripInteraction_2_sum_cbf not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor AtomUpdate_2_sum not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor TripInteraction_3_had_rbf not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor TripInteraction_3_sum_cbf not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor AtomUpdate_3_sum not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor OutBlock_2_sum not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor OutBlock_2_had not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor OutBlock_3_sum not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/scaling/compat.py:77: (WARNING): Scale factor OutBlock_3_had not found in model
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:568: (INFO): Loaded GemNetT with 3360519 parameters.
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/logger.py:175: (WARNING): log_summary for Tensorboard not supported
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:358: (INFO): Loading dataset: trajectory_lmdb
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/train_100')]'
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f2423bb8200>, batch_size=16, drop_last=False
/opt/hostedtoolcache/Python/3.12.9/x64/lib/python3.12/site-packages/torch/utils/data/dataloader.py:557: UserWarning: This DataLoader will create 8 worker processes in total. Our suggested max number of worker in current system is 4, which is smaller than what this DataLoader is going to create. Please be aware that excessive worker creation might get DataLoader running slow or even freeze, lower the worker number to avoid potential slowness/freeze if necessary.
warnings.warn(_create_warning_msg(
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/datasets/base_dataset.py:94: (WARNING): Could not find dataset metadata.npz files in '[PosixPath('data/s2ef/val_20')]'
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:166: (WARNING): Disabled BalancedBatchSampler because num_replicas=1.
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:179: (WARNING): Failed to get data sizes, falling back to uniform partitioning. BalancedBatchSampler requires a dataset that has a metadata attributed with number of atoms.
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:88: (INFO): rank: 0: Sampler created...
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/data_parallel.py:200: (INFO): Created BalancedBatchSampler with sampler=<fairchem.core.common.data_parallel.StatefulDistributedSampler object at 0x7f245d242f30>, batch_size=8, drop_last=False
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy'] have been saved to: ./checkpoints/2025-03-29-04-52-16-S2EF-gemnet-t/normalizers.pt
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/_load_utils.py:112: (INFO): normalizers checkpoint for targets ['energy', 'forces'] have been saved to: ./checkpoints/2025-03-29-04-52-16-S2EF-gemnet-t/normalizers.pt
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output energy: mean=-0.7554450631141663, rmsd=2.887317180633545.
2025-03-29 04:52:26 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:491: (INFO): Normalization values for output forces: mean=0.0, rmsd=2.887317180633545.
2025-03-29 04:53:00 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.05e+02, forcesx_mae: 3.30e+00, forcesy_mae: 4.12e+00, forcesz_mae: 3.19e+00, forces_mae: 3.54e+00, forces_cosine_similarity: -1.29e-02, forces_magnitude_error: 6.83e+00, loss: 2.84e+02, lr: 1.00e-04, epoch: 7.14e-01, step: 5.00e+00
2025-03-29 04:53:08 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 2.20it/s]
device 0: 67%|██████▋ | 2/3 [00:00<00:00, 3.19it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 4.02it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 3.12it/s]
2025-03-29 04:53:10 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 2.1859, forcesx_mae: 2.3018, forcesy_mae: 1.8443, forcesz_mae: 1.9866, forces_mae: 2.0442, forces_cosine_similarity: -0.1886, forces_magnitude_error: 3.8822, loss: 146.1000, epoch: 1.0000
2025-03-29 04:53:32 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 3.40e+01, forcesx_mae: 1.67e+00, forcesy_mae: 2.28e+00, forcesz_mae: 1.70e+00, forces_mae: 1.88e+00, forces_cosine_similarity: -6.95e-03, forces_magnitude_error: 3.54e+00, loss: 1.38e+02, lr: 1.00e-04, epoch: 1.43e+00, step: 1.00e+01
2025-03-29 04:53:53 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 2.55it/s]
device 0: 67%|██████▋ | 2/3 [00:00<00:00, 3.35it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 4.07it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 3.26it/s]
2025-03-29 04:53:54 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 2.8872, forcesx_mae: 2.2966, forcesy_mae: 1.8156, forcesz_mae: 1.9075, forces_mae: 2.0066, forces_cosine_similarity: -0.1919, forces_magnitude_error: 3.7918, loss: 143.2075, epoch: 2.0000
2025-03-29 04:54:02 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 4.95e+01, forcesx_mae: 1.18e+00, forcesy_mae: 1.38e+00, forcesz_mae: 1.18e+00, forces_mae: 1.25e+00, forces_cosine_similarity: 1.80e-02, forces_magnitude_error: 2.22e+00, loss: 9.82e+01, lr: 1.00e-04, epoch: 2.14e+00, step: 1.50e+01
2025-03-29 04:54:35 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 5.73e+01, forcesx_mae: 8.89e-01, forcesy_mae: 1.20e+00, forcesz_mae: 9.26e-01, forces_mae: 1.01e+00, forces_cosine_similarity: -2.94e-02, forces_magnitude_error: 1.74e+00, loss: 9.04e+01, lr: 1.00e-04, epoch: 2.86e+00, step: 2.00e+01
2025-03-29 04:54:37 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 2.12it/s]
device 0: 67%|██████▋ | 2/3 [00:00<00:00, 3.08it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 3.95it/s]
device 0: 100%|██████████| 3/3 [00:01<00:00, 3.00it/s]
2025-03-29 04:54:38 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 1.6403, forcesx_mae: 2.2450, forcesy_mae: 1.7636, forcesz_mae: 1.8652, forces_mae: 1.9579, forces_cosine_similarity: -0.1887, forces_magnitude_error: 3.6963, loss: 139.6124, epoch: 3.0000
2025-03-29 04:55:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 2.06e+01, forcesx_mae: 6.35e-01, forcesy_mae: 7.63e-01, forcesz_mae: 6.23e-01, forces_mae: 6.74e-01, forces_cosine_similarity: 4.42e-02, forces_magnitude_error: 1.12e+00, loss: 5.37e+01, lr: 1.00e-04, epoch: 3.57e+00, step: 2.50e+01
2025-03-29 04:55:20 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 2.14it/s]
device 0: 67%|██████▋ | 2/3 [00:00<00:00, 3.12it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 3.92it/s]
device 0: 100%|██████████| 3/3 [00:01<00:00, 2.94it/s]
2025-03-29 04:55:22 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 2.1157, forcesx_mae: 2.2045, forcesy_mae: 1.7296, forcesz_mae: 1.8392, forces_mae: 1.9244, forces_cosine_similarity: -0.1851, forces_magnitude_error: 3.6256, loss: 137.3321, epoch: 4.0000
2025-03-29 04:55:36 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.13e+01, forcesx_mae: 5.82e-01, forcesy_mae: 8.45e-01, forcesz_mae: 6.18e-01, forces_mae: 6.82e-01, forces_cosine_similarity: 5.76e-02, forces_magnitude_error: 1.08e+00, loss: 5.14e+01, lr: 1.00e-04, epoch: 4.29e+00, step: 3.00e+01
2025-03-29 04:56:04 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/ocp_trainer.py:194: (INFO): energy_forces_within_threshold: 0.00e+00, energy_mae: 1.02e+01, forcesx_mae: 4.69e-01, forcesy_mae: 6.54e-01, forcesz_mae: 5.04e-01, forces_mae: 5.42e-01, forces_cosine_similarity: 3.08e-02, forces_magnitude_error: 8.93e-01, loss: 4.17e+01, lr: 1.00e-04, epoch: 5.00e+00, step: 3.50e+01
2025-03-29 04:56:04 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:863: (INFO): Evaluating on val.
device 0: 0%| | 0/3 [00:00<?, ?it/s]
device 0: 33%|███▎ | 1/3 [00:00<00:00, 2.48it/s]
device 0: 67%|██████▋ | 2/3 [00:00<00:00, 3.26it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 3.95it/s]
device 0: 100%|██████████| 3/3 [00:00<00:00, 3.06it/s]
2025-03-29 04:56:05 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:905: (INFO): energy_forces_within_threshold: 0.0000, energy_mae: 1.9943, forcesx_mae: 2.1967, forcesy_mae: 1.7273, forcesz_mae: 1.8153, forces_mae: 1.9131, forces_cosine_similarity: -0.1857, forces_magnitude_error: 3.6056, loss: 136.5640, epoch: 5.0000
This model should achieve a force MAE of 0.0668, a force cosine of 0.1180, and an energy MAE of 0.8106 in 2 epochs, significantly better than our simple model.
Again, we encourage the reader to try playing with no. of blocks, choice of basis functions, the various embedding sizes to develop intuition for the interplay between these hyperparameters.
(Optional) OCP Calculator #
For those interested in using our pretrained models for other applications, we provide an [ASE](https://wiki.fysik.dtu.dk/ase/#:~:text=The%20Atomic%20Simulation%20Environment%20(ASE,under%20the%20GNU%20LGPL%20license.)-compatible Calculator to interface with ASE’s functionality.
Download pretrained checkpoint#
We have released checkpoints of all the models on the leaderboard here. These trained models can be used as an ASE calculator for various calculations.
For this tutorial we download one of our earlier model checkpoints: GemNet-T
from fairchem.core.models.model_registry import model_name_to_local_file
checkpoint_path = model_name_to_local_file('GemNet-dT-S2EF-OC20-All', local_cache='/tmp/fairchem_checkpoints/')
2025-03-29 04:56:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/models/model_registry.py:59: (INFO): Checking local cache: /tmp/fairchem_checkpoints/ for model GemNet-dT-S2EF-OC20-All
Using the OCP Calculator#
from fairchem.core.common.relaxation.ase_utils import OCPCalculator
import ase.io
from ase.optimize import BFGS
from ase.build import fcc100, add_adsorbate, molecule
import os
from ase.constraints import FixAtoms
# Construct a sample structure
adslab = fcc100("Cu", size=(3, 3, 3))
adsorbate = molecule("C3H8")
add_adsorbate(adslab, adsorbate, 3, offset=(1, 1))
tags = np.zeros(len(adslab))
tags[18:27] = 1
tags[27:] = 2
adslab.set_tags(tags)
cons= FixAtoms(indices=[atom.index for atom in adslab if (atom.tag == 0)])
adslab.set_constraint(cons)
adslab.center(vacuum=13.0, axis=2)
adslab.set_pbc(True)
# Define the calculator
calc = OCPCalculator(checkpoint_path=checkpoint_path)
# Set up the calculator
adslab.calc = calc
os.makedirs("data/sample_ml_relax", exist_ok=True)
opt = BFGS(adslab, trajectory="data/sample_ml_relax/toy_c3h8_relax.traj")
opt.run(fmax=0.05, steps=100)
/home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/ase_utils.py:200: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
checkpoint = torch.load(checkpoint_path, map_location=torch.device("cpu"))
2025-03-29 04:56:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/utils.py:1319: (WARNING): Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
2025-03-29 04:56:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:208: (INFO): amp: false
cmd:
checkpoint_dir: /home/runner/work/fairchem/fairchem/docs/legacy_tutorials/checkpoints/2025-03-29-04-56-32
commit: core:b88b66e,experimental:NA
identifier: ''
logs_dir: /home/runner/work/fairchem/fairchem/docs/legacy_tutorials/logs/wandb/2025-03-29-04-56-32
print_every: 100
results_dir: /home/runner/work/fairchem/fairchem/docs/legacy_tutorials/results/2025-03-29-04-56-32
seed: null
timestamp_id: 2025-03-29-04-56-32
version: 0.1.dev1+gb88b66e
dataset:
format: trajectory_lmdb
grad_target_mean: 0.0
grad_target_std: 2.887317180633545
key_mapping:
force: forces
y: energy
normalize_labels: true
target_mean: -0.7554450631141663
target_std: 2.887317180633545
transforms:
normalizer:
energy:
mean: -0.7554450631141663
stdev: 2.887317180633545
forces:
mean: 0.0
stdev: 2.887317180633545
evaluation_metrics:
metrics:
energy:
- mae
forces:
- forcesx_mae
- forcesy_mae
- forcesz_mae
- mae
- cosine_similarity
- magnitude_error
misc:
- energy_forces_within_threshold
gp_gpus: null
gpus: 0
logger: wandb
loss_functions:
- energy:
coefficient: 1
fn: mae
- forces:
coefficient: 100
fn: l2mae
model:
activation: silu
cbf:
name: spherical_harmonics
cutoff: 6.0
direct_forces: true
emb_size_atom: 512
emb_size_bil_trip: 64
emb_size_cbf: 16
emb_size_edge: 512
emb_size_rbf: 16
emb_size_trip: 64
envelope:
exponent: 5
name: polynomial
extensive: true
max_neighbors: 50
name: gemnet_t
num_after_skip: 2
num_atom: 3
num_before_skip: 1
num_blocks: 3
num_concat: 1
num_radial: 128
num_spherical: 7
otf_graph: true
output_init: HeOrthogonal
rbf:
name: gaussian
regress_forces: true
optim:
batch_size: 32
clip_grad_norm: 10
ema_decay: 0.999
energy_coefficient: 1
eval_batch_size: 32
eval_every: 5000
factor: 0.8
force_coefficient: 100
loss_energy: mae
loss_force: l2mae
lr_initial: 0.0005
max_epochs: 80
mode: min
num_workers: 2
optimizer: AdamW
optimizer_params:
amsgrad: true
patience: 3
scheduler: ReduceLROnPlateau
outputs:
energy:
level: system
forces:
eval_on_free_atoms: true
level: atom
train_on_free_atoms: true
relax_dataset: {}
slurm: {}
task:
dataset: trajectory_lmdb
description: Regressing to energies and forces for DFT trajectories from OCP
eval_on_free_atoms: true
grad_input: atomic forces
labels:
- potential energy
metric: mae
train_on_free_atoms: true
type: regression
test_dataset: {}
trainer: ocp
val_dataset: {}
2025-03-29 04:56:06 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:557: (INFO): Loading model: gemnet_t
2025-03-29 04:56:07 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:568: (INFO): Loaded GemNetT with 31671825 parameters.
2025-03-29 04:56:07 /home/runner/work/fairchem/fairchem/src/fairchem/core/trainers/base_trainer.py:621: (INFO): Loading checkpoint in inference-only mode, not loading keys associated with trainer state!
/home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/normalizer.py:69: UserWarning: To copy construct from a tensor, it is recommended to use sourceTensor.clone().detach() or sourceTensor.clone().detach().requires_grad_(True), rather than torch.tensor(sourceTensor).
"mean": torch.tensor(state_dict["mean"]),
2025-03-29 04:56:07 /home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/ase_utils.py:258: (WARNING): No seed has been set in modelcheckpoint or OCPCalculator! Results may not be reproducible on re-run
Step Time Energy fmax
BFGS: 0 04:56:08 -4.099784 1.567516
BFGS: 1 04:56:08 -4.244472 1.136985
BFGS: 2 04:56:09 -4.403122 0.763561
BFGS: 3 04:56:09 -4.503644 0.836383
BFGS: 4 04:56:10 -4.558209 0.733877
BFGS: 5 04:56:10 -4.592066 0.409535
BFGS: 6 04:56:11 -4.619359 0.731256
BFGS: 7 04:56:11 -4.671464 0.971141
BFGS: 8 04:56:12 -4.796473 0.921026
BFGS: 9 04:56:13 -4.957972 0.976196
BFGS: 10 04:56:13 -5.109419 1.038484
BFGS: 11 04:56:14 -5.295610 1.224973
BFGS: 12 04:56:14 -5.498995 1.127049
BFGS: 13 04:56:15 -5.618104 1.066863
BFGS: 14 04:56:15 -5.737133 0.950848
BFGS: 15 04:56:16 -5.901944 0.926049
BFGS: 16 04:56:16 -6.076124 1.273782
BFGS: 17 04:56:17 -6.198373 1.202949
BFGS: 18 04:56:17 -6.250327 0.685198
BFGS: 19 04:56:18 -6.254099 0.200784
BFGS: 20 04:56:18 -6.293955 0.177882
BFGS: 21 04:56:19 -6.326336 0.229420
BFGS: 22 04:56:19 -6.324459 0.170037
BFGS: 23 04:56:20 -6.321297 0.101550
BFGS: 24 04:56:20 -6.328354 0.084693
BFGS: 25 04:56:21 -6.331687 0.058673
BFGS: 26 04:56:21 -6.331860 0.044462
True
(Optional) Creating your own LMDBs for use in the OCP repository#
In order to interface with our repository, the data mustbe structured and organized in a specific format. Below we walk you through on how to create such datasets with your own non-OC20 data that may help with your research.
For this tutorial we use the toy C3H8 trajectory we previously generated here.
Initial Structure to Relaxed Energy (IS2RE) LMDBs#
IS2RE/IS2RS LMDBs utilize the LmdbDataset dataset. This dataset expects the data to be contained in a single LMDB file. In addition to the attributes defined by AtomsToGraph, the following attributes must be added for the IS2RE/IS2RS tasks:
pos_relaxed: Relaxed adslab positions
sid: Unique system identifier, arbitrary
y_init: Initial adslab energy, formerly Data.y
y_relaxed: Relaxed adslab energy
tags (optional): 0 - subsurface, 1 - surface, 2 - adsorbate
As a demo, we will use the above generated data to create an IS2R* LMDB file.
from fairchem.core.preprocessing import AtomsToGraphs
"""
args description:
max neigh (int): maximum number of neighors to be considered while constructing a graph
radius (int): Neighbors are considered only within this radius cutoff in Angstrom
r_energy (bool): Stored energy value in the Data object; False for test data
r_forces (bool): Stores forces value in the Data object; False for test data
r_distances (bool): pre-calculates distances taking into account PBC and max neigh/radius
If you set it to False, make sure to add "otf_graph = True" under models in config for runs
r_fixed (bools): True if you want to fix the subsurface atoms
"""
a2g = AtomsToGraphs(
max_neigh=50,
radius=6,
r_energy=True,
r_forces=True,
r_distances=False,
r_fixed=True,
)
import lmdb
"""
For most cases one just needs to change the name of the lmdb as they require.
Make sure to give the entire path in the config (with .lmdb) for IS2RE tasks
"""
db = lmdb.open(
"data/toy_C3H8.lmdb",
map_size=1099511627776 * 2,
subdir=False,
meminit=False,
map_async=True,
)
"""
This method converts extracts all features from trajectory file and convert to Data Object
"""
def read_trajectory_extract_features(a2g, traj_path):
# Read the traj file
traj = ase.io.read(traj_path, ":")
# Get tags if you had defined those in the atoms object, if not skip this line
tags = traj[0].get_tags()
# Collect only initial and final image as this is IS2RS task
images = [traj[0], traj[-1]]
# Converts a list of atoms object to a list of Data object using a2g defined above
data_objects = a2g.convert_all(images, disable_tqdm=True)
# Add tags to the data objects if you have them (we would suggest to do so), if not skip this
data_objects[0].tags = torch.LongTensor(tags)
data_objects[1].tags = torch.LongTensor(tags)
return data_objects
import torch
import pickle
system_paths = ["data/toy_c3h8_relax.traj"] # specify list of trajectory files you wish to write to LMDBs
idx = 0
for system in system_paths:
# Extract Data object
data_objects = read_trajectory_extract_features(a2g, system)
initial_struc = data_objects[0]
relaxed_struc = data_objects[1]
initial_struc.y_init = initial_struc.y # subtract off reference energy, if applicable
del initial_struc.y
initial_struc.y_relaxed = relaxed_struc.y # subtract off reference energy, if applicable
initial_struc.pos_relaxed = relaxed_struc.pos
# Filter data if necessary
# OCP filters adsorption energies > |10| eV
initial_struc.sid = idx # arbitrary unique identifier
# no neighbor edge case check
if initial_struc.edge_index.shape[1] == 0:
print("no neighbors", traj_path)
continue
# Write to LMDB
txn = db.begin(write=True)
txn.put(f"{idx}".encode("ascii"), pickle.dumps(initial_struc, protocol=-1))
txn.commit()
db.sync()
idx += 1
db.close()
from fairchem.core.datasets import LmdbDataset
# LmdbDataset is out custom Dataset method to read the lmdbs as Data objects. Note that we need to give the entire path (including lmdb) for IS2RE
dataset = LmdbDataset({"src": "data/toy_C3H8.lmdb"})
print("Size of the dataset created:", len(dataset))
print(dataset[0])
Size of the dataset created: 1
Data(pos=[38, 3], cell=[1, 3, 3], atomic_numbers=[38], natoms=38, tags=[38], edge_index=[2, 1733], cell_offsets=[1733, 3], edge_distance_vec=[1733, 3], nedges=1733, energy=15.804699620275976, forces=[38, 3], fixed=[38], pos_relaxed=[38, 3], sid=0)
Structure to Energy and Forces (S2EF) LMDBs#
S2EF LMDBs utilize the LmdbDatset dataset. This dataset expects a directory of LMDB files. In addition to the attributes defined by AtomsToGraph, the following attributes must be added for the S2EF task:
tags (optional): 0 - subsurface, 1 - surface, 2 - adsorbate
fid: Frame index along the trajcetory
sid- sid: Unique system identifier, arbitrary
Additionally, a “length” key must be added to each LMDB file.
As a demo, we will use the above generated data to create an S2EF LMDB dataset
os.makedirs("data/s2ef", exist_ok=True)
db = lmdb.open(
"data/s2ef/toy_C3H8.lmdb",
map_size=1099511627776 * 2,
subdir=False,
meminit=False,
map_async=True,
)
from tqdm import tqdm
tags = traj[0].get_tags()
data_objects = a2g.convert_all(traj, disable_tqdm=True)
for fid, data in tqdm(enumerate(data_objects), total=len(data_objects)):
#assign sid
data.sid = torch.LongTensor([0])
#assign fid
data.fid = torch.LongTensor([fid])
#assign tags, if available
data.tags = torch.LongTensor(tags)
# Filter data if necessary
# OCP filters adsorption energies > |10| eV and forces > |50| eV/A
# no neighbor edge case check
if data.edge_index.shape[1] == 0:
print("no neighbors", traj_path)
continue
txn = db.begin(write=True)
txn.put(f"{fid}".encode("ascii"), pickle.dumps(data, protocol=-1))
txn.commit()
txn = db.begin(write=True)
txn.put(f"length".encode("ascii"), pickle.dumps(len(data_objects), protocol=-1))
txn.commit()
db.sync()
db.close()
0%| | 0/101 [00:00<?, ?it/s]
25%|██▍ | 25/101 [00:00<00:00, 249.53it/s]
83%|████████▎ | 84/101 [00:00<00:00, 448.26it/s]
100%|██████████| 101/101 [00:00<00:00, 441.00it/s]
Running on command line [Preferred way to train models] #
The previous sections of this notebook are intended to demonstrate the inner workings of our codebase. For regular training, we suggest that you train and evaluate on command line.
Clone our repo at https://github.com/FAIR-Chem/fairchem and set up the environment according to the readme.
Download relevant data (see above for info).
In the config file, modify the path of the data train val, normalization parameters as well as any other model or training args.
For a simple example, we’ll train DimeNet++ on IS2RE demo data:
a. Modify the train data path in /contents/ocp/configs/oc20/is2re/10k/base.yml in
Line 4 to /contents/ocp/data/is2re/train_10k/data.lmdb and val data path in Line 8 to /contents/ocp/data/is2re/val_2k/data.lmdb.
b. Calculate the mean and std for train data and modify Lines 6-7 respectively
c. We can change the model parameters in /contents/ocp/configs/oc20/is2re/10k/dimenet_plus_plus/dpp.yml and we suggest you to change the lr_milestones and warmup_steps as the data here is smaller (these need to be tuned for every dataset).
Train:
python main.py --mode train --config-yml configs/oc20/is2re/10k/dimenet_plus_plus/dpp.yml --identifier dpp_is2re_sample
# Optional block to try command line training
# Note that config args can be added in the command line. For example, --optim.batch_size=1
Add a data path as a test set to
configs/oc20/is2re/10k/base.ymlRun predictions with the trained model:
python main.py --mode predict --config-yml configs/oc20/is2re/10k/dimenet_plus_plus/dpp.yml --checkpoint checkpoints/[datetime]-dpp_is2re_sample/checkpoint.ptView energy predictions at
results/[datetime]/is2re_predictions.npz
For more information on how to train and evaluate, see this readme. For checkpoints of publicly available trained models, see MODELS.md.
Limitations #
The OpenCatalyst project is motivated by the problems we face due to climate change, many of which require innovative solutions to reduce energy usage and replace traditional chemical feedstocks with renewable alternatives. For example, one of the most energy intensive chemical processes is the development of new electrochemical catalysts for ammonia fertilizer production that helped to feed the world’s growing population during the 20th century. This is also an illustrative example of possible unintended consequences as advancements in chemistry and materials may be used for numerous purposes. As ammonia fertilization increased in use, its overuse in today’s farming has led to ocean “dead zones” and its production is very carbon intensive. Knowledge and techniques used to create ammonia were also transferred to the creation of explosives during wartime. We hope to steer the use of ML for atomic simulations to societally-beneficial uses by training and testing our approaches on datasets, such as OC20, that were specifically designed to address chemical reactions useful for addressing climate change.
Next Steps #
While progress has been well underway - https://opencatalystproject.org/leaderboard.html, a considerable gap still exists between state-of-the-art models and our target goals. We offer some some general thoughts as to next steps for the readers to ponder on or explore:
GNN depth has consistenly improved model performance. What limitations to depth are there? How far can we push deeper models for OC20?
Our best performing models have little to no physical biases encoded. Can we incorporate such biases to improve our models? Experiments with physically inspired embeddings have had no advantage vs. random initializations, are there better ways to incorporate this information into the models?
Uncertainty estimation will play an important role in later stages of the project when it comes to large scale screening. How can we get reliable uncertainty estimates from large scale GNNs?
Are we limited to message-passing GNNs? Can we leverage alternative architectures for similiar or better performance?
Trajectories are nothing more than sequential data points. How can we use sequential modeling techniques to model the full trajectory?
OC20 is a large and diverse dataset with many splits. For those with limited resources but unsure where to start, we provide some general recommendations:
The IS2RE-direct task is a great place to start. With the largest training set containing ~460k data points, this task is easily accesible for those with even just a single GPU.
Those interested in the more general S2EF task don’t need to train on the All set to get meaningful performance.
Results on the 2M dataset are often sufficient to highlight model improvements.
For a fixed compute budget (e.g. fixed number of steps), training on the All set often leads to better performance.
The S2EF 200k dataset is fairly noisy, trying to find meaningful trends using this dataset can be difficult.
References#
Open Catalyst codebase: https://github.com/FAIR-Chem/fairchem/
Open Catalyst webpage: https://opencatalystproject.org/
Electrocatalysis white paper: C. Lawrence Zitnick, Lowik Chanussot, Abhishek Das, Siddharth Goyal, Javier Heras-Domingo, Caleb Ho, Weihua Hu, Thibaut Lavril, Aini Palizhati, Morgane Riviere, Muhammed Shuaibi, Anuroop Sriram, Kevin Tran, Brandon Wood, Junwoong Yoon, Devi Parikh, Zachary Ulissi: “An Introduction to Electrocatalyst Design using Machine Learning for Renewable Energy Storage”, 2020; arXiv:2010.09435.
OC20 dataset paper: L. Chanussot, A. Das, S. Goyal, T. Lavril, M. Shuaibi, M. Riviere, K. Tran, J. Heras-Domingo, C. Ho, W. Hu, A. Palizhati, A. Sriram, B. Wood, J. Yoon, D. Parikh, C. L. Zitnick, and Z. Ulissi. The Open Catalyst 2020 (oc20) dataset and community challenges. ACS Catalysis, 2021.
Gemnet model: Johannes Klicpera, Florian Becker, and Stephan Günnemann. Gemnet: Universal directional graph neural networks for molecules, 2021.