Simple simulations using the OCP ASE calculator#
To introduce OCP we start with using it to calculate adsorption energies for a simple, atomic adsorbate where we specify the site we want to the adsorption energy for. Conceptually, you do this like you would do it with density functional theory. You create a slab model for the surface, place an adsorbate on it as an initial guess, run a relaxation to get the lowest energy geometry, and then compute the adsorption energy using reference states for the adsorbate.
You do have to be careful in the details though. Some OCP model/checkpoint combinations return a total energy like density functional theory would, but some return an “adsorption energy” directly. You have to know which one you are using. In this example, the model we use returns an “adsorption energy”.
Calculating adsorption energies#
Calculating adsorption energy with OCP Adsorption energies are different than you might be used to in OCP. For example, you may want the adsorption energy of oxygen, and conventionally you would compute that from this reaction:
1/2 O2 + slab -> slab-O
This is not what is done in OCP. It is referenced to a different reaction
x CO + (x + y/2 - z) H2 + (z-x) H2O + w/2 N2 + * -> CxHyOzNw*
Here, x=y=w=0, z=1, so the reaction ends up as
-H2 + H2O + * -> O*
or alternatively,
H2O + * -> O* + H2
It is possible through thermodynamic cycles to compute other reactions. If we can look up rH1 below and compute rH2
H2 + 1/2 O2 -> H2O re1 = -3.03 eV
H2O + * -> O* + H2 re2 # Get from OCP as a direct calculation
Then, the adsorption energy for
1/2O2 + * -> O*
is just re1 + re2.
Based on https://atct.anl.gov/Thermochemical%20Data/version%201.118/species/?species_number=986, the formation energy of water is about -3.03 eV at standard state. You could also compute this using DFT.
The first step is getting a checkpoint for the model we want to use. eSCN is currently the state of the art model arXiv. This next cell will download the checkpoint if you don’t have it already. However, we’re going to use an older GemNet model (which still works pretty well!) just to keep the resources lower for this tutorial.
The different models have different compute requirements. If you find your kernel is crashing, it probably means you have exceeded the allowed amount of memory. This checkpoint works fine in this example, but it may crash your kernel if you use it in the NRR example.
from fairchem.core.models.model_registry import model_name_to_local_file
checkpoint_path = model_name_to_local_file('EquiformerV2-31M-S2EF-OC20-All+MD', local_cache='/tmp/fairchem_checkpoints/')
Next we load the checkpoint. The output is somewhat verbose, but it can be informative for debugging purposes.
from fairchem.core.common.relaxation.ase_utils import OCPCalculator
calc = OCPCalculator(checkpoint_path=checkpoint_path, cpu=False)
# calc = OCPCalculator(checkpoint_path=checkpoint_path, cpu=True)
/home/runner/work/fairchem/fairchem/src/fairchem/core/models/scn/spherical_harmonics.py:23: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
_Jd = torch.load(os.path.join(os.path.dirname(__file__), "Jd.pt"))
/home/runner/work/fairchem/fairchem/src/fairchem/core/models/equiformer_v2/wigner.py:10: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
_Jd = torch.load(os.path.join(os.path.dirname(__file__), "Jd.pt"))
/home/runner/work/fairchem/fairchem/src/fairchem/core/models/escn/so3.py:23: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
_Jd = torch.load(os.path.join(os.path.dirname(__file__), "Jd.pt"))
/home/runner/work/fairchem/fairchem/src/fairchem/core/common/relaxation/ase_utils.py:200: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
checkpoint = torch.load(checkpoint_path, map_location=torch.device("cpu"))
WARNING:root:Detected old config, converting to new format. Consider updating to avoid potential incompatibilities.
INFO:root:amp: true
cmd:
checkpoint_dir: /home/runner/work/fairchem/fairchem/docs/tutorials/checkpoints/2025-03-29-05-09-20
commit: core:b88b66e,experimental:NA
identifier: ''
logs_dir: /home/runner/work/fairchem/fairchem/docs/tutorials/logs/wandb/2025-03-29-05-09-20
print_every: 100
results_dir: /home/runner/work/fairchem/fairchem/docs/tutorials/results/2025-03-29-05-09-20
seed: null
timestamp_id: 2025-03-29-05-09-20
version: 0.1.dev1+gb88b66e
dataset:
format: trajectory_lmdb_v2
grad_target_mean: 0.0
grad_target_std: 2.887317180633545
key_mapping:
force: forces
y: energy
normalize_labels: true
target_mean: -0.7554450631141663
target_std: 2.887317180633545
transforms:
normalizer:
energy:
mean: -0.7554450631141663
stdev: 2.887317180633545
forces:
mean: 0.0
stdev: 2.887317180633545
evaluation_metrics:
metrics:
energy:
- mae
forces:
- forcesx_mae
- forcesy_mae
- forcesz_mae
- mae
- cosine_similarity
- magnitude_error
misc:
- energy_forces_within_threshold
primary_metric: forces_mae
gp_gpus: null
gpus: 0
logger: wandb
loss_functions:
- energy:
coefficient: 4
fn: mae
- forces:
coefficient: 100
fn: l2mae
model:
alpha_drop: 0.1
attn_activation: silu
attn_alpha_channels: 64
attn_hidden_channels: 64
attn_value_channels: 16
distance_function: gaussian
drop_path_rate: 0.1
edge_channels: 128
ffn_activation: silu
ffn_hidden_channels: 128
grid_resolution: 18
lmax_list:
- 4
max_neighbors: 20
max_num_elements: 90
max_radius: 12.0
mmax_list:
- 2
name: equiformer_v2
norm_type: layer_norm_sh
num_distance_basis: 512
num_heads: 8
num_layers: 8
num_sphere_samples: 128
otf_graph: true
proj_drop: 0.0
regress_forces: true
sphere_channels: 128
use_atom_edge_embedding: true
use_gate_act: false
use_grid_mlp: true
use_pbc: true
use_s2_act_attn: false
weight_init: uniform
optim:
batch_size: 8
clip_grad_norm: 100
ema_decay: 0.999
energy_coefficient: 4
eval_batch_size: 8
eval_every: 10000
force_coefficient: 100
grad_accumulation_steps: 1
load_balancing: atoms
loss_energy: mae
loss_force: l2mae
lr_initial: 0.0004
max_epochs: 3
num_workers: 8
optimizer: AdamW
optimizer_params:
weight_decay: 0.001
scheduler: LambdaLR
scheduler_params:
epochs: 1009275
lambda_type: cosine
lr: 0.0004
lr_min_factor: 0.01
warmup_epochs: 3364.25
warmup_factor: 0.2
outputs:
energy:
level: system
forces:
eval_on_free_atoms: true
level: atom
train_on_free_atoms: true
relax_dataset: {}
slurm:
additional_parameters:
constraint: volta32gb
cpus_per_task: 9
folder: /checkpoint/abhshkdz/open-catalyst-project/logs/equiformer_v2/8307793
gpus_per_node: 8
job_id: '8307793'
job_name: eq2s_051701_allmd
mem: 480GB
nodes: 8
ntasks_per_node: 8
partition: learnaccel
time: 4320
task:
dataset: trajectory_lmdb_v2
eval_on_free_atoms: true
grad_input: atomic forces
labels:
- potential energy
primary_metric: forces_mae
train_on_free_atoms: true
test_dataset: {}
trainer: ocp
val_dataset: {}
INFO:root:Loading model: equiformer_v2
WARNING:root:equiformer_v2 (EquiformerV2) class is deprecated in favor of equiformer_v2_backbone_and_heads (EquiformerV2BackboneAndHeads)
INFO:root:Loaded EquiformerV2 with 31058690 parameters.
INFO:root:Loading checkpoint in inference-only mode, not loading keys associated with trainer state!
/home/runner/work/fairchem/fairchem/src/fairchem/core/modules/normalization/normalizer.py:69: UserWarning: To copy construct from a tensor, it is recommended to use sourceTensor.clone().detach() or sourceTensor.clone().detach().requires_grad_(True), rather than torch.tensor(sourceTensor).
"mean": torch.tensor(state_dict["mean"]),
WARNING:root:No seed has been set in modelcheckpoint or OCPCalculator! Results may not be reproducible on re-run
Next we can build a slab with an adsorbate on it. Here we use the ASE module to build a Pt slab. We use the experimental lattice constant that is the default. This can introduce some small errors with DFT since the lattice constant can differ by a few percent, and it is common to use DFT lattice constants. In this example, we do not constrain any layers.
from ase.build import fcc111, add_adsorbate
from ase.optimize import BFGS
re1 = -3.03
slab = fcc111('Pt', size=(2, 2, 5), vacuum=10.0)
add_adsorbate(slab, 'O', height=1.2, position='fcc')
slab.set_calculator(calc)
opt = BFGS(slab)
opt.run(fmax=0.05, steps=100)
slab_e = slab.get_potential_energy()
slab_e + re1
/tmp/ipykernel_5437/3494085424.py:6: FutureWarning: Please use atoms.calc = calc
slab.set_calculator(calc)
Step Time Energy fmax
BFGS: 0 05:09:51 1.212762 1.631530
BFGS: 1 05:09:52 1.071970 0.906285
BFGS: 2 05:09:53 0.981418 0.637568
BFGS: 3 05:09:53 0.960160 0.689581
BFGS: 4 05:09:54 0.881589 0.561222
BFGS: 5 05:09:55 0.835982 0.376775
BFGS: 6 05:09:55 0.824551 0.420583
BFGS: 7 05:09:56 0.814235 0.512939
BFGS: 8 05:09:57 0.827347 0.902106
BFGS: 9 05:09:57 0.779738 0.358875
BFGS: 10 05:09:58 0.772442 0.206212
BFGS: 11 05:09:59 0.757418 0.122774
BFGS: 12 05:09:59 0.755282 0.112772
BFGS: 13 05:10:00 0.754121 0.105044
BFGS: 14 05:10:01 0.755008 0.087029
BFGS: 15 05:10:01 0.757414 0.047153
-2.2725855994224546
It is good practice to look at your geometries to make sure they are what you expect.
import matplotlib.pyplot as plt
from ase.visualize.plot import plot_atoms
fig, axs = plt.subplots(1, 2)
plot_atoms(slab, axs[0]);
plot_atoms(slab, axs[1], rotation=('-90x'))
axs[0].set_axis_off()
axs[1].set_axis_off()
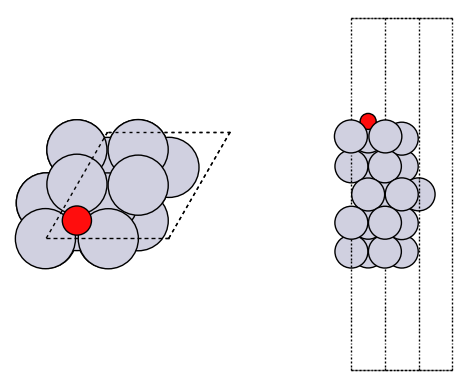
How did we do? We need a reference point. In the paper below, there is an atomic adsorption energy for O on Pt(111) of about -4.264 eV. This is for the reaction O + * -> O*. To convert this to the dissociative adsorption energy, we have to add the reaction:
1/2 O2 -> O D = 2.58 eV (expt)
to get a comparable energy of about -1.68 eV. There is about ~0.6 eV difference (we predicted -2.3 eV above, and the reference comparison is -1.68 eV) to account for. The biggest difference is likely due to the differences in exchange-correlation functional. The reference data used the PBE functional, and eSCN was trained on RPBE data. To additional places where there are differences include:
Difference in lattice constant
The reference energy used for the experiment references. These can differ by up to 0.5 eV from comparable DFT calculations.
How many layers are relaxed in the calculation
Some of these differences tend to be systematic, and you can calibrate and correct these, especially if you can augment these with your own DFT calculations.
See convergence study for some additional studies of factors that influence this number.
Exercises#
Explore the effect of the lattice constant on the adsorption energy.
Try different sites, including the bridge and top sites. Compare the energies, and inspect the resulting geometries.
Trends in adsorption energies across metals.#
Xu, Z., & Kitchin, J. R. (2014). Probing the coverage dependence of site and adsorbate configurational correlations on (111) surfaces of late transition metals. J. Phys. Chem. C, 118(44), 25597–25602. http://dx.doi.org/10.1021/jp508805h
These are atomic adsorption energies:
O + * -> O*
We have to do some work to get comparable numbers from OCP
H2 + 1/2 O2 -> H2O re1 = -3.03 eV
H2O + * -> O* + H2 re2 # Get from OCP
O -> 1/2 O2 re3 = -2.58 eV
Then, the adsorption energy for
O + * -> O*
is just re1 + re2 + re3.
Here we just look at the fcc site on Pt. First, we get the data stored in the paper.
Next we get the structures and compute their energies. Some subtle points are that we have to account for stoichiometry, and normalize the adsorption energy by the number of oxygens.
First we get a reference energy from the paper (PBE, 0.25 ML O on Pt(111)).
import json
with open('energies.json') as f:
edata = json.load(f)
with open('structures.json') as f:
sdata = json.load(f)
edata['Pt']['O']['fcc']['0.25']
-4.263842000000002
Next, we load data from the SI to get the geometry to start from.
with open('structures.json') as f:
s = json.load(f)
sfcc = s['Pt']['O']['fcc']['0.25']
Next, we construct the atomic geometry, run the geometry optimization, and compute the energy.
re3 = -2.58 # O -> 1/2 O2 re3 = -2.58 eV
from ase import Atoms
atoms = Atoms(sfcc['symbols'],
positions=sfcc['pos'],
cell=sfcc['cell'],
pbc=True)
atoms.set_calculator(calc)
opt = BFGS(atoms)
opt.run(fmax=0.05, steps=100)
re2 = atoms.get_potential_energy()
nO = 0
for atom in atoms:
if atom.symbol == 'O':
nO += 1
re2 += re1 + re3
print(re2 / nO)
/tmp/ipykernel_5437/2476004300.py:10: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calc)
Step Time Energy fmax
BFGS: 0 05:10:03 0.720968 0.217973
BFGS: 1 05:10:03 0.717579 0.182151
BFGS: 2 05:10:04 0.707525 0.067372
BFGS: 3 05:10:05 0.705303 0.062100
BFGS: 4 05:10:05 0.701208 0.049768
-4.90879206418991
Site correlations#
This cell reproduces a portion of a figure in the paper. We compare oxygen adsorption energies in the fcc and hcp sites across metals and coverages. These adsorption energies are highly correlated with each other because the adsorption sites are so similar.
At higher coverages, the agreement is not as good. This is likely because the model is extrapolating and needs to be fine-tuned.
from tqdm import tqdm
import time
t0 = time.time()
data = {'fcc': [],
'hcp': []}
refdata = {'fcc': [],
'hcp': []}
for metal in ['Cu', 'Ag', 'Pd', 'Pt', 'Rh', 'Ir']:
print(metal)
for site in ['fcc', 'hcp']:
for adsorbate in ['O']:
for coverage in tqdm(['0.25']):
entry = s[metal][adsorbate][site][coverage]
atoms = Atoms(entry['symbols'],
positions=entry['pos'],
cell=entry['cell'],
pbc=True)
atoms.set_calculator(calc)
opt = BFGS(atoms, logfile=None) # no logfile to suppress output
opt.run(fmax=0.05, steps=100)
re2 = atoms.get_potential_energy()
nO = 0
for atom in atoms:
if atom.symbol == 'O':
nO += 1
re2 += re1 + re3
data[site] += [re2 / nO]
refdata[site] += [edata[metal][adsorbate][site][coverage]]
f'Elapsed time = {time.time() - t0} seconds'
Cu
0%| | 0/1 [00:00<?, ?it/s]
/tmp/ipykernel_5437/1169188772.py:27: FutureWarning: Please use atoms.calc = calc
atoms.set_calculator(calc)
100%|██████████| 1/1 [00:03<00:00, 3.27s/it]
100%|██████████| 1/1 [00:03<00:00, 3.27s/it]
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:03<00:00, 3.79s/it]
100%|██████████| 1/1 [00:03<00:00, 3.79s/it]
Ag
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:02<00:00, 2.16s/it]
100%|██████████| 1/1 [00:02<00:00, 2.16s/it]
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:03<00:00, 3.80s/it]
100%|██████████| 1/1 [00:03<00:00, 3.80s/it]
Pd
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:03<00:00, 3.29s/it]
100%|██████████| 1/1 [00:03<00:00, 3.29s/it]
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:03<00:00, 3.81s/it]
100%|██████████| 1/1 [00:03<00:00, 3.81s/it]
Pt
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:02<00:00, 2.70s/it]
100%|██████████| 1/1 [00:02<00:00, 2.70s/it]
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:01<00:00, 1.62s/it]
100%|██████████| 1/1 [00:01<00:00, 1.62s/it]
Rh
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:00<00:00, 1.84it/s]
100%|██████████| 1/1 [00:00<00:00, 1.84it/s]
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:00<00:00, 1.85it/s]
100%|██████████| 1/1 [00:00<00:00, 1.85it/s]
Ir
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:01<00:00, 1.70s/it]
100%|██████████| 1/1 [00:01<00:00, 1.70s/it]
0%| | 0/1 [00:00<?, ?it/s]
100%|██████████| 1/1 [00:01<00:00, 1.10s/it]
100%|██████████| 1/1 [00:01<00:00, 1.10s/it]
'Elapsed time = 28.350666522979736 seconds'
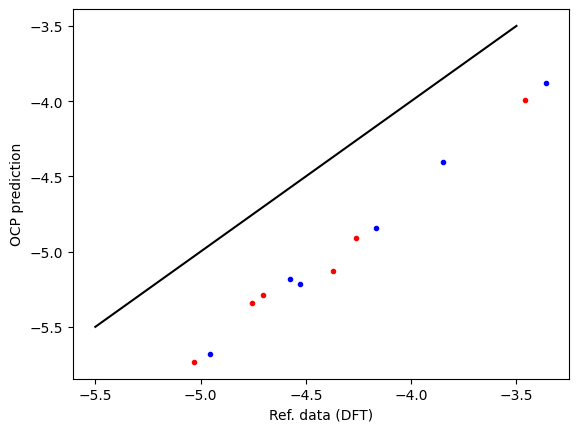
First, we compare the computed data and reference data. There is a systematic difference of about 0.5 eV due to the difference between RPBE and PBE functionals, and other subtle differences like lattice constant differences and reference energy differences. This is pretty typical, and an expected deviation.
plt.plot(refdata['fcc'], data['fcc'], 'r.', label='fcc')
plt.plot(refdata['hcp'], data['hcp'], 'b.', label='hcp')
plt.plot([-5.5, -3.5], [-5.5, -3.5], 'k-')
plt.xlabel('Ref. data (DFT)')
plt.ylabel('OCP prediction');
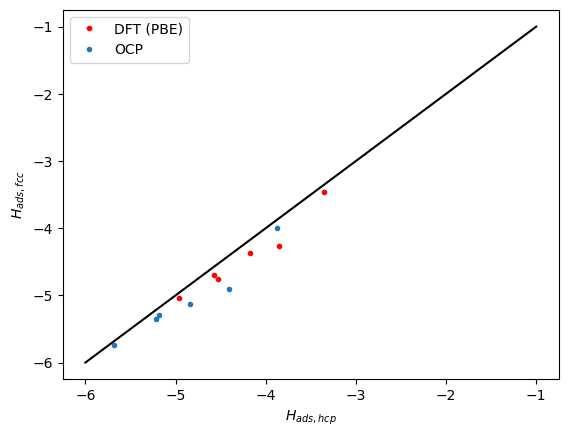
Next we compare the correlation between the hcp and fcc sites. Here we see the same trends. The data falls below the parity line because the hcp sites tend to be a little weaker binding than the fcc sites.
plt.plot(refdata['hcp'], refdata['fcc'], 'r.')
plt.plot(data['hcp'], data['fcc'], '.')
plt.plot([-6, -1], [-6, -1], 'k-')
plt.xlabel('$H_{ads, hcp}$')
plt.ylabel('$H_{ads, fcc}$')
plt.legend(['DFT (PBE)', 'OCP']);
Exercises#
You can also explore a few other adsorbates: C, H, N.
Explore the higher coverages. The deviations from the reference data are expected to be higher, but relative differences tend to be better. You probably need fine tuning to improve this performance. This data set doesn’t have forces though, so it isn’t practical to do it here.
Next steps#
In the next step, we consider some more complex adsorbates in nitrogen reduction, and how we can leverage OCP to automate the search for the most stable adsorbate geometry. See the next step.
Convergence study#
In Calculating adsorption energies we discussed some possible reasons we might see a discrepancy. Here we investigate some factors that impact the computed energies.
In this section, the energies refer to the reaction 1/2 O2 -> O*.
Effects of number of layers#
Slab thickness could be a factor. Here we relax the whole slab, and see by about 4 layers the energy is converged to ~0.02 eV.
for nlayers in [3, 4, 5, 6, 7, 8]:
slab = fcc111('Pt', size=(2, 2, nlayers), vacuum=10.0)
add_adsorbate(slab, 'O', height=1.2, position='fcc')
slab.set_calculator(calc)
opt = BFGS(slab, logfile=None)
opt.run(fmax=0.05, steps=100)
slab_e = slab.get_potential_energy()
print(f'nlayers = {nlayers}: {slab_e + re1:1.2f} eV')
/tmp/ipykernel_5437/4214609233.py:5: FutureWarning: Please use atoms.calc = calc
slab.set_calculator(calc)
nlayers = 3: -2.38 eV
nlayers = 4: -2.26 eV
nlayers = 5: -2.27 eV
nlayers = 6: -2.26 eV
nlayers = 7: -2.26 eV
nlayers = 8: -2.27 eV
Effects of relaxation#
It is common to only relax a few layers, and constrain lower layers to bulk coordinates. We do that here. We only relax the adsorbate and the top layer.
This has a small effect (0.1 eV).
from ase.constraints import FixAtoms
for nlayers in [3, 4, 5, 6, 7, 8]:
slab = fcc111('Pt', size=(2, 2, nlayers), vacuum=10.0)
add_adsorbate(slab, 'O', height=1.2, position='fcc')
slab.set_constraint(FixAtoms(mask=[atom.tag > 1 for atom in slab]))
slab.set_calculator(calc)
opt = BFGS(slab, logfile=None)
opt.run(fmax=0.05, steps=100)
slab_e = slab.get_potential_energy()
print(f'nlayers = {nlayers}: {slab_e + re1:1.2f} eV')
/tmp/ipykernel_5437/2738246227.py:9: FutureWarning: Please use atoms.calc = calc
slab.set_calculator(calc)
nlayers = 3: -2.22 eV
nlayers = 4: -2.07 eV
nlayers = 5: -2.09 eV
nlayers = 6: -2.08 eV
nlayers = 7: -2.09 eV
nlayers = 8: -2.10 eV
Unit cell size#
Coverage effects are quite noticeable with oxygen. Here we consider larger unit cells. This effect is large, and the results don’t look right, usually adsorption energies get more favorable at lower coverage, not less. This suggests fine-tuning could be important even at low coverages.
for size in [1, 2, 3, 4, 5]:
slab = fcc111('Pt', size=(size, size, 5), vacuum=10.0)
add_adsorbate(slab, 'O', height=1.2, position='fcc')
slab.set_constraint(FixAtoms(mask=[atom.tag > 1 for atom in slab]))
slab.set_calculator(calc)
opt = BFGS(slab, logfile=None)
opt.run(fmax=0.05, steps=100)
slab_e = slab.get_potential_energy()
print(f'({size}x{size}): {slab_e + re1:1.2f} eV')
/tmp/ipykernel_5437/1073633840.py:7: FutureWarning: Please use atoms.calc = calc
slab.set_calculator(calc)
(1x1): -1.00 eV
(2x2): -2.09 eV
(3x3): -1.44 eV
(4x4): -1.48 eV
(5x5): -1.37 eV
Summary#
As with DFT, you should take care to see how these kinds of decisions affect your results, and determine if they would change any interpretations or not.